Abordagem
O passo mais importante é a coleta minuciosa da história.
Identificação dos fatores de risco
Os fatores de risco fortemente associados à doença de Huntington incluem a presença de história familiar positiva e a presença de repetição de citosina-adenina-guanina (CAG) expandida na região N-terminal do gene da huntingtina. Ocasionalmente, não é encontrada história familiar da doença de Huntington. Nesses casos, deve-se considerar falsa paternidade, óbito precoce de um dos pais afetados ou expansão da repetição de CAG de um dos pais não afetados.
Comprometimento cognitivo
Os primeiros sintomas da doença de Huntington estão relacionados à perda de funções executivas, que tipicamente são mais bem percebidas em discussões sobre o trabalho ou a vida doméstica do paciente. Os achados comuns incluem:
Comprometimento da concentração (o desempenho no trabalho ou na escola pode estar comprometido)
Apatia e ansiedade na realização de tarefas
Aumento de erros em funções complexas
Apropriação de responsabilidades por colegas ou membros da família
Escorregões, julgamentos errôneos ou acidentes com veículo automotor.
Embora não considerado um aspecto típico do exame físico, a comparação com parceiros ou irmãos com níveis similares de educação ou quociente de inteligência (QI) geralmente é informativa.
Características comportamentais
As manifestações comportamentais comumente incluem:[25]
Irritabilidade
Impulsividade
Falta de atenção à aparência ou à higiene
Alterações da personalidade. Embora às vezes sejam difíceis de categorizar, geralmente são percebidas como alterações nos hábitos e nos interesses. Se houver negação, pode ser útil entrevistar os membros da família separadamente.[7][26]
Frequentemente, os pacientes parecem um pouco desinibidos ou excepcionalmente ansiosos. Testes formais do estado mental à beira do leito, como o mini-exame do estado mental, geralmente são normais, especialmente se o paciente tiver boa escolaridade. O reconhecimento de inadequação ocasional ou a ausência de foco do paciente é mais útil, especialmente em comparação com o parceiro ou com o membro da família que o acompanha.
Características motoras
As características motoras podem incluir:
Coreia
Distonia
Espasmos/agitação: os membros da família podem notar a presença dessas características e de alterações na coordenação
Bradicinesia e rigidez.
Os achados mais úteis no exame físico estão relacionados à coreia.[7][26][27] Os pacientes às vezes são capazes de suprimir movimentos involuntários, portanto é útil passar alguns minutos procurando especificamente por eles. Pode ser útil fazer isso com o paciente sentado, sem os sapatos, de olhos fechados, realizando o teste dos sete seriados com as mãos estendidas. Movimentos aleatórios dos dedos das mãos (por exemplo, agitar os dedos) e dos pés, com posturas peculiares ocasionais das mãos, do tronco ou dos membros e expressões faciais estranhas são típicos da doença de Huntington precoce, assim como o ritmo e a frequência irregulares da percussão do dedo indicador no polegar (“finger tapping”), além da impersistência motora (incapacidade de manter a língua totalmente protruída).
A demonstração da coreia em termos verbais é difícil, mas uma eletromiografia (EMG) de um músculo do dedo da mão em um paciente afetado demonstra a natureza aleatória desse tipo de distúrbio do movimento. Pode ser detectada uma lentidão no movimento ocular rápido (movimento sacádico). Essa característica geralmente é proeminente na doença de Huntington juvenil. Também pode ser observada dificuldade na marcha em tandem.
Para médicos que têm experiência limitada com coreia e doença de Huntington, recomenda-se o encaminhamento a um especialista em distúrbios do movimento ou a um centro médico envolvido com pesquisa sobre a doença de Huntington.[Figure caption and citation for the preceding image starts]: Eletromiografia (EMG) da coreia no estágio I da doença; registro feito com eletrodos de superfície padrão tipo disco, no ventre muscular/tendão, sobre o primeiro músculo interósseo dorsalWalker FO. Huntington's disease. Lancet 2007 Jan 20;369(9557):218-28. Usado com permissão [Citation ends].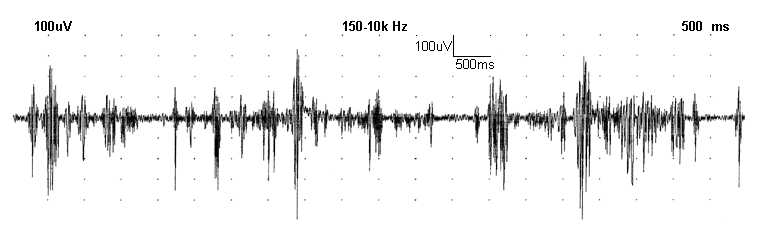
Outras características
A depressão pode ser uma característica comórbida da doença, mas é inespecífica e não é uma manifestação inicial comum.[25][28][29][30] Podem ocorrer comportamentos obsessivos e compulsões, mas, novamente, em geral não estão presentes até uma fase mais tardia da doença, frequentemente após o diagnóstico ser feito.[25]
Perturbações do sono são comuns e podem se tornar mais graves, à medida que a doença evolui.[31] A perturbação do sono na doença de Huntington pode tomar a forma de latência do sono aumentada (início do sono tardio), eficácia do sono reduzida (dificuldade de manter o sono), despertar precoce, ciclos diurno-noturno prejudicados, sonolência diurna excessiva ou ritmo circadiano prejudicado.[32] A perturbação do sono também pode ser causada por comorbidades, como ansiedade ou depressão.
Exames por imagem
Os exames de imagem geralmente não são úteis na avaliação ou diagnóstico da doença de Huntington precoce. Entretanto, a atrofia do núcleo caudado, aparente na tomografia computadorizada (TC) ou na RNM, é um achado consistente em estágios tardios da doença.[19]
Consentimento para exames adicionais
O diagnóstico é útil para a documentação da incapacidade, mas pode ser indesejável devido à ausência de uma terapia modificadora da doença. Pode ser útil informar os pacientes sobre as potenciais implicações de um diagnóstico na família e em planos de saúde, informar que eles apresentam características que podem ou não ser evidências da doença de Huntington e perguntar se eles gostariam de ser encaminhados ao aconselhamento genético pré-teste. O teste genético diagnóstico pode ser considerado para um indivíduo sintomático em uma variedade de cenários e, em cada caso, as consequências e implicações de todos os desfechos possíveis devem ser cuidadosamente considerados antes de proceder.[33] Esse teste pode ser adiado a critério do paciente e pode ajudá-lo a decidir como gerenciar questões em andamento sobre a vida, as finanças ou o trabalho.[7][26]
Teste genético
Se o paciente tiver características típicas da doença de Huntington, o teste confirmatório mais útil será o teste de repetição de CAG. Ele normalmente demora algumas semanas. Um resultado positivo tem muitas implicações para o paciente e para a família, então geralmente é melhor para o paciente ser encaminhado para o aconselhamento genético ou geneticista clínico, para que receba informações adequadas sobre a doença de Huntington antecipadamente, para que esteja preparado. Ao apresentar os resultados de um teste positivo, geralmente se providencia que o paciente venha com seu parceiro ou um amigo ou membro da família que possa apoiá-lo. A notícia do diagnóstico pode ser um evento emocional significativo para o paciente e representa um período importante na educação da família sobre a doença de Huntington e suas implicações genéticas para membros da família e para o planejamento familiar. Se a testagem genética confirmatória for negativa, é provável que o paciente precise ser encaminhado a um especialista em distúrbios do movimento para detectar outras possíveis causas para seus sintomas.
A testagem também pode ser realizada em pacientes idosos em risco, sem sintomas da doença de Huntington, para definir o risco da doença em seus descendentes. Recomenda-se a realização de testes genéticos pré-sintomáticos para membros da família em risco apenas em centros médicos especializados em tais testes, utilizando-se diretrizes publicadas. Testes preditivos para pacientes com menos de 18 anos não são recomendados; os pacientes que solicitarem testes preditivos devem ter acesso a aconselhamento genético.[34][35][36]
[Figure caption and citation for the preceding image starts]: Ciclo de vida na doença de Huntington (DH)Walker FO. Huntington's disease. Lancet 2007 Jan 20;369(9557):218-28. Usado com permissão [Citation ends].
O uso deste conteúdo está sujeito ao nosso aviso legal