Deve-se suspeitar de talassemia alfa em pacientes com a origem étnica apropriada e com microcitose sem evidência de deficiência de ferro, com ou sem anemia concomitante. As manifestações clínicas são amplamente variáveis, desde assintomática até, raramente, dependência de transfusão.
A avaliação inicial deve se concentrar na anamnese e no exame físico; os exames laboratoriais iniciais devem incluir um hemograma completo com índices eritrocitários, contagem de reticulócitos e uma análise cuidadosa do esfregaço do sangue periférico.[3]Vichinsky E. Complexity of alpha thalassemia: growing health problem with new approaches to screening, diagnosis, and therapy. Ann N Y Acad Sci. 2010 Aug;1202:180-7.
http://www.ncbi.nlm.nih.gov/pubmed/20712791?tool=bestpractice.com
História
Os pacientes com 1 ou 2 genes da globina alfa afetados provavelmente serão assintomáticos.
Os pacientes com doença da hemoglobina (Hb) H são variavelmente sintomáticos. Em um estudo realizado em pacientes de Hong Kong com Hb H, apenas 24% dos que tinham Hb H delecional apresentaram sintomas, em comparação com 40% dos que tinham doença de Hb H não delecional.[5]Chen FE, Ooi C, Ha SY, et al. Genetic and clinical features of hemoglobin H disease in Chinese patients. N Engl J Med. 2000 Aug 24;343(8):544-50.
https://www.nejm.org/doi/full/10.1056/NEJM200008243430804
http://www.ncbi.nlm.nih.gov/pubmed/10954762?tool=bestpractice.com
A história deve incluir o seguinte:
Presença e duração de sintomas relacionados à anemia (fadiga, dispneia, tontura)
Presença e duração de sintomas relacionados à icterícia (descoloração amarela da esclera, da pele e das membranas mucosas)
Presença e duração de sintomas relacionados aos cálculos biliares (náuseas, flatulência, distensão abdominal e dor abdominal)
História prévia de suplementação de ferro ou transfusão de eritrócitos (embora a maioria dos pacientes com doença de Hb H não necessite de transfusões crônicas, em um estudo até um terço dos pacientes com Hb H não delecional necessitaram de transfusões regulares)[8]Vichinsky EP, MacKlin EA, Waye JS, et al. Changes in the epidemiology of thalassemia in North America: a new minority disease. Pediatrics. 2005 Dec;116(6):e818-25.
http://www.ncbi.nlm.nih.gov/pubmed/16291734?tool=bestpractice.com
Origens étnicas do paciente (África Subsaariana, Bacia Mediterrânea, Oriente Médio, Subcontinente Indiano e Sudeste Asiático)
História de outros membros da família afetados
Idade do paciente (uma vez que a talassemia alfa pode ter uma variabilidade tão grande nas manifestações clínicas, os pacientes podem apresentar desde hidropisia fetal no período intrauterino a microcitose assintomática em qualquer instante da vida adulta; no entanto, aqueles com manifestações mais graves geralmente apresentam as manifestações clínicas na infância ou no início da vida adulta).
Exame físico
O exame físico pode estar normal. Na doença da Hb H, pode ocorrer icterícia, e a esplenomegalia é um achado comum, também podendo ser observada hepatomegalia[5]Chen FE, Ooi C, Ha SY, et al. Genetic and clinical features of hemoglobin H disease in Chinese patients. N Engl J Med. 2000 Aug 24;343(8):544-50.
https://www.nejm.org/doi/full/10.1056/NEJM200008243430804
http://www.ncbi.nlm.nih.gov/pubmed/10954762?tool=bestpractice.com
[46]Chui DH, Fucharoen S, Chan V. Hemoglobin H disease: not necessarily a benign disorder. Blood. 2003 Feb 1;101(3):791-800.
https://ashpublications.org/blood/article/101/3/791/88824/Hemoglobin-H-disease-not-necessarily-a-benign
http://www.ncbi.nlm.nih.gov/pubmed/12393486?tool=bestpractice.com
Aspectos clínicos de anemia podem estar presentes, com sintomas que incluem fadiga, tontura e dispneia. Sintomas de cálculos biliares (distensão abdominal, dor abdominal e flatulência) podem estar presentes.
Podem ocorrer alterações esqueléticas devido à expansão das células da linhagem eritroide da medula óssea, com uma massa óssea baixa relatada em alguns pacientes.[47]Wiromrat P, Rattanathongkom A, Laoaroon N, et al. Bone mineral density and Dickkopf-1 in adolescents with non-deletional hemoglobin H disease. J Clin Densitom. 2023 Apr 26;101379.
http://www.ncbi.nlm.nih.gov/pubmed/37147222?tool=bestpractice.com
Raramente, alterações esqueléticas podem acarretar uma apresentação mais leve dos dismorfismos faciais observados na talassemia beta maior tratada de forma inadequada, com hipertrofia do maxilar, bossa frontal e proeminência das eminências malares.[46]Chui DH, Fucharoen S, Chan V. Hemoglobin H disease: not necessarily a benign disorder. Blood. 2003 Feb 1;101(3):791-800.
https://ashpublications.org/blood/article/101/3/791/88824/Hemoglobin-H-disease-not-necessarily-a-benign
http://www.ncbi.nlm.nih.gov/pubmed/12393486?tool=bestpractice.com
[48]Laosombat V, Viprakasit V, Chotsampancharoen T, et al. Clinical features and molecular analysis in Thai patients with HbH disease. Ann Hematol. 2009 Dec;88(12):1185-92.
http://www.ncbi.nlm.nih.gov/pubmed/19390853?tool=bestpractice.com
Também pode ser observado retardo do crescimento nas crianças.[5]Chen FE, Ooi C, Ha SY, et al. Genetic and clinical features of hemoglobin H disease in Chinese patients. N Engl J Med. 2000 Aug 24;343(8):544-50.
https://www.nejm.org/doi/full/10.1056/NEJM200008243430804
http://www.ncbi.nlm.nih.gov/pubmed/10954762?tool=bestpractice.com
[48]Laosombat V, Viprakasit V, Chotsampancharoen T, et al. Clinical features and molecular analysis in Thai patients with HbH disease. Ann Hematol. 2009 Dec;88(12):1185-92.
http://www.ncbi.nlm.nih.gov/pubmed/19390853?tool=bestpractice.com
Tem sido descrita hematopoese extramedular ocasionando massas paraespinhais.[49]Wu JH, Shih LY, Kuo TT, et al. Intrathoracic extramedullary hematopoietic tumor in hemoglobin H disease. Am J Hematol. 1992 Dec;41(4):285-8.
http://www.ncbi.nlm.nih.gov/pubmed/1288291?tool=bestpractice.com
[50]Benz EJ Jr, Wu CC, Sohani AR. Case records of the Massachusetts General Hospital. Case 25-2011. A 62-year-old woman with anemia and paraspinal masses. N Engl J Med. 2011 Aug 18;365(7):648-58.
http://www.ncbi.nlm.nih.gov/pubmed/21848466?tool=bestpractice.com
Avaliação laboratorial inicial
A avaliação laboratorial inicial deve incluir hemograma completo, contagem de reticulócitos, testes hemolíticos e índices de eritrócitos, incluindo medição do volume corpuscular médio (VCM), hemoglobina corpuscular média (HCM) e contagem de eritrócitos.[16]Thalassaemia International Federation. Guidelines for the management of alpha-thalassaemia (2023). 2023 [internet publication].
https://thalassaemia.org.cy/publications/tif-publications/guidelines-for-the-management-of-%ce%b1-thalassaemia
Caso haja microcitose (VCM <78 fentolitros) ou hipocromia (HCM <27 picogramas/célula), deverá ser avaliado o estado do ferro (ferro sérico, transferrina, saturação de transferrina e ferritina) para diferenciar entre anemia decorrente de talassemia alfa e anemia ferropriva.[16]Thalassaemia International Federation. Guidelines for the management of alpha-thalassaemia (2023). 2023 [internet publication].
https://thalassaemia.org.cy/publications/tif-publications/guidelines-for-the-management-of-%ce%b1-thalassaemia
Se os resultados do perfil de ferro forem ambíguos ou limítrofes, pode-se justificar uma tentativa curta, bem-monitorada de suplementação de ferro para descartar anemia ferropriva.
O esfregaço de sangue periférico deve ser cuidadosamente analisado quanto à ocorrência de achados consistentes com talassemia alfa, incluindo microcitose, hipocromia, aumento da policromasia, células-alvo e anisopoiquilocitose.[16]Thalassaemia International Federation. Guidelines for the management of alpha-thalassaemia (2023). 2023 [internet publication].
https://thalassaemia.org.cy/publications/tif-publications/guidelines-for-the-management-of-%ce%b1-thalassaemia
Na doença da Hb H, podem ser encontrados eritrócitos deformados e até mesmo fragmentados, podendo ser observados corpos de inclusão característicos com um corante supravital, como o azul de cresil brilhante.[16]Thalassaemia International Federation. Guidelines for the management of alpha-thalassaemia (2023). 2023 [internet publication].
https://thalassaemia.org.cy/publications/tif-publications/guidelines-for-the-management-of-%ce%b1-thalassaemia
[Figure caption and citation for the preceding image starts]: Doença da hemoglobina HDo acervo de Elizabeth A. Price e Stanley L. Schrier, Stanford University [Citation ends].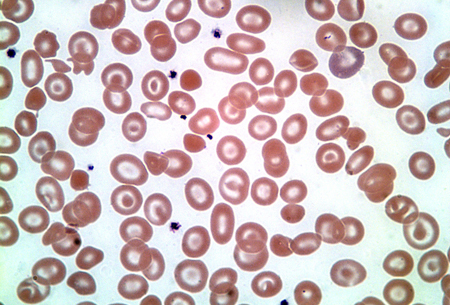
Na doença de Hb H, a porcentagem de reticulócitos está elevada (2.5% a 4.0% para doença da Hb H delecional; 5.5% a 6.5% para doença da Hb H não delecional) e pode aumentar ainda mais durante infecções agudas ou crises hemolíticas.[16]Thalassaemia International Federation. Guidelines for the management of alpha-thalassaemia (2023). 2023 [internet publication].
https://thalassaemia.org.cy/publications/tif-publications/guidelines-for-the-management-of-%ce%b1-thalassaemia
Na “Hb Constant Spring” (alongamento da globina alfa) homozigótica, as células estarão normais ou de tamanho ligeiramente diminuído e o pontilhado basofílico pode estar proeminente.[6]Pootrakul P, Winichagoon P, Fucharoen S, et al. Homozygous haemoglobin Constant Spring: a need for revision of concept. Hum Genet. 1981;59(3):250-5.
http://www.ncbi.nlm.nih.gov/pubmed/7327587?tool=bestpractice.com
Avaliação laboratorial subsequente
Hb H e Hb Bart podem ser detectadas como hemoglobinas de rápida evolução. A Hb H nem sempre é detectável de forma confiável pela eletroforese da hemoglobinas de rotina, e alguns especialistas acreditam que os corpos de inclusão da Hb H sejam mais confiáveis para o diagnóstico da doença da Hb H.[4]Harteveld CL, Higgs DR. Alpha-thalassemia. Orphanet J Rare Dis. 2010 May 28;5:13.
https://ojrd.biomedcentral.com/articles/10.1186/1750-1172-5-13
http://www.ncbi.nlm.nih.gov/pubmed/20507641?tool=bestpractice.com
O fracionamento e a medição automática da Hb também podem ser realizados por meio de cromatografia líquida de alta eficiência (HPLC).[4]Harteveld CL, Higgs DR. Alpha-thalassemia. Orphanet J Rare Dis. 2010 May 28;5:13.
https://ojrd.biomedcentral.com/articles/10.1186/1750-1172-5-13
http://www.ncbi.nlm.nih.gov/pubmed/20507641?tool=bestpractice.com
[16]Thalassaemia International Federation. Guidelines for the management of alpha-thalassaemia (2023). 2023 [internet publication].
https://thalassaemia.org.cy/publications/tif-publications/guidelines-for-the-management-of-%ce%b1-thalassaemia
Essas modalidades de teste supostamente identificarão também todos os distúrbios comuns da hemoglobina (ou seja, Hb E, Hb S, HB C, Hb D) que podem estar presentes e podem afetar a evolução clínica. No entanto, a eletroforese da Hb e a HPLC não detectarão deleções ou mutações em apenas um ou dois genes da globina alfa, nem diferenciarão a doença da Hb H delecional da não delecional (exceto para a “Hb Constant Spring” [alongamento da globina alfa]).
Caracterização da talassemia alfa
Sempre requer testes genéticos de globina alfa baseados em DNA. Sete das deleções de talassemia alfa mais comuns (-alfa (3.7), -alfa (4.2), --(FIL), --(THAI), --(MED), -(alfa) (20.5), --(SEA)) podem ser diagnosticadas por reação em cadeia da polimerase de gap (gap-PCR).[16]Thalassaemia International Federation. Guidelines for the management of alpha-thalassaemia (2023). 2023 [internet publication].
https://thalassaemia.org.cy/publications/tif-publications/guidelines-for-the-management-of-%ce%b1-thalassaemia
Outros alelos de deleção são detectados por amplificação de múltiplas sondas dependentes de ligação.[16]Thalassaemia International Federation. Guidelines for the management of alpha-thalassaemia (2023). 2023 [internet publication].
https://thalassaemia.org.cy/publications/tif-publications/guidelines-for-the-management-of-%ce%b1-thalassaemia
Mutações de talassemia alfa não delecionais geralmente são detectadas por sequenciamento direto ou dot-blot reverso.[16]Thalassaemia International Federation. Guidelines for the management of alpha-thalassaemia (2023). 2023 [internet publication].
https://thalassaemia.org.cy/publications/tif-publications/guidelines-for-the-management-of-%ce%b1-thalassaemia
[51]Sabath DE. Molecular diagnosis of thalassemias and hemoglobinopathies: an ACLPS critical review. Am J Clin Pathol. 2017 Jul 1;148(1):6-15.
https://academic.oup.com/ajcp/article/148/1/6/3866692
http://www.ncbi.nlm.nih.gov/pubmed/28605432?tool=bestpractice.com
[52]Chan V, Yam I, Chen FE, et al. A reverse dot-blot method for rapid detection of non-deletion alpha thalassaemia. Br J Haematol. 1999 Mar;104(3):513-5.
http://www.ncbi.nlm.nih.gov/pubmed/10086788?tool=bestpractice.com
[53]Vijian D, Wan Ab Rahman WS, Ponnuraj KT, et al. Molecular detection of alpha thalassemia: a review of prevalent techniques. Medeni Med J. 2021;36(3):257-69.
https://pmc.ncbi.nlm.nih.gov/articles/PMC8565582
http://www.ncbi.nlm.nih.gov/pubmed/34915685?tool=bestpractice.com
Não existe uma abordagem simples para detectar todas as mutações conhecidas. Podem ser necessários laboratórios de referência com experiência no diagnóstico de hemoglobinopatias para diagnosticar casos difíceis de forma oportuna, particularmente com a finalidade de aconselhamento genético.
Sobrecarga de ferro
Se os níveis de ferritina sérica sugerirem estado de ferro elevado, a sobrecarga de ferro no fígado deve ser avaliada por ressonância nuclear magnética (RNM) R2 ou R2*, por dispositivos supercondutores de interferência quântica (SQUID) ou por biópsia hepática (menos preferencial).[16]Thalassaemia International Federation. Guidelines for the management of alpha-thalassaemia (2023). 2023 [internet publication].
https://thalassaemia.org.cy/publications/tif-publications/guidelines-for-the-management-of-%ce%b1-thalassaemia
[54]Northern California Comprehensive Thalassemia Center. Standards of care guidelines for thalassemia. 2012 [internet publication].
https://thalassemia.ucsf.edu/standards-care
[55]Wood JC. Diagnosis and management of transfusion iron overload: the role of imaging. Am J Hematol. 2007 Dec;82(suppl 12):1132-5.
https://onlinelibrary.wiley.com/doi/epdf/10.1002/ajh.21099
http://www.ncbi.nlm.nih.gov/pubmed/17963249?tool=bestpractice.com
Os níveis de ferritina sérica podem subestimar a concentração hepática de ferro.[56]Lal A, Goldrich ML, Haines DA, et al. Heterogeneity of hemoglobin H disease in childhood. N Engl J Med. 2011 Feb 24;364(8):710-8.
https://www.nejm.org/doi/full/10.1056/NEJMoa1010174
http://www.ncbi.nlm.nih.gov/pubmed/21345100?tool=bestpractice.com
As diretrizes indicam a avaliação por RNM da concentração hepática de ferro se a ferritina sérica estiver >674.1 picomoles/L (>300 nanogramas/mL) em pacientes com doença da Hb H.[16]Thalassaemia International Federation. Guidelines for the management of alpha-thalassaemia (2023). 2023 [internet publication].
https://thalassaemia.org.cy/publications/tif-publications/guidelines-for-the-management-of-%ce%b1-thalassaemia
A carga cardíaca de ferro é avaliada por RNM cardíaca em T2*.[16]Thalassaemia International Federation. Guidelines for the management of alpha-thalassaemia (2023). 2023 [internet publication].
https://thalassaemia.org.cy/publications/tif-publications/guidelines-for-the-management-of-%ce%b1-thalassaemia
A carga de ferro cardíaca é incomum em pacientes não submetidos a transfusão.