Etiologia
A hemoglobina humana consiste em um tetrâmero de 2 pares de cadeias de globina: 1 par de cadeias do tipo alfa e 1 par de cadeias não alfa, cada um contendo um grupo heme, e possui a função essencial de levar oxigênio a todas as partes do corpo, a partir dos pulmões. O grupo de genes da globina alfa em cada cromossomo 16 contém 1 gene embriônico zeta e 2 genes da globina alfa coexpressos, alfa-2 e alfa-1. Normalmente, o gene alfa-2, localizado a montante do gene alfa-1, codifica 2 a 3 vezes mais proteínas que o gene alfa-1.[24]
Existem 2 variedades principais de talassemia alfa: talassemia alfa(0) (--/), em que ambos os genes da globina alfa no mesmo cromossomo sofrem deleção, e talassemia alfa(+) (-alpha/), em que apenas 1 dos 2 genes da globina alfa no mesmo cromossomo sofre deleção ou mutação. Em geral, as variantes não delecionais possuem um fenótipo mais intenso que as variantes delecionais mais comuns.[5] Aparentemente, isso se deve principalmente a um gene alfa-2 com mutação interferindo na capacidade do gene alfa-1 aumentar a produção.[1]
As manifestações clínicas também são influenciadas pela presença ou não de defeitos concomitantes na síntese da cadeia de globina beta. Deve-se observar que portadores de genes alfa multiplicados (ou seja, cópias extras de genes da globina alfa) são assintomáticos.
Fisiopatologia
A talassemia alfa é caracterizada pela produção diminuída ou ausente de, pelo menos, 1 dos 4 genes da globina alfa, e o fenótipo clínico corresponde ao grau de deficiência na síntese da cadeia de globina alfa, o que, de maneira similar, se correlaciona amplamente com o genótipo. Portanto, a fisiopatologia depende, em um pequeno grau, do grau de deficiência na formação da hemoglobina (Hb), mas em alto grau ela decorre do acúmulo do excesso de cadeias de globina beta não emparelhadas, as quais se agregam, causando danos oxidativos e mecânicos aos eritrócitos afetados e acarretando sua destruição prematura.
Os aspectos clínicos da talassemia alfa são predominantemente aqueles associados à anemia e ao aumento da hemólise.
A síntese reduzida de cadeia de globina alfa e a diminuição de Hb A (alfa 2 beta 2) causa a microcitose e a hipocromia características observadas com a doença. Além disso, a produção diminuída de globina alfa ocasiona um excesso de cadeias não alfa livres e a formação de tetrâmeros gama 4 (Hb Bart) e beta 4 (Hb H). A Hb Bart e a Hb H possuem alta afinidade com o oxigênio, não apresentam interação heme-heme ou tampouco efeito Bohr, não sendo, portanto, funcionais como portadoras de oxigênio.[25] As manifestações clínicas dependem do grau de deficiência da síntese da cadeia de globina alfa e, portanto, se correlacionarão ao defeito genético.
A hemólise, com destruição predominantemente extravascular, é observada na hemoglobina H (Hb H) e na Hb H/Constant Spring, mas também na doença homozigótica Hb Constant Spring (alfa(CS)alfa/alfa(CS)alfa).[6][26][27][28] Os eritrócitos Hb H e Hb H/Constant Spring são anormalmente rígidos, possivelmente em decorrência do aumento das associações entre a globina e as membranas dos eritrócitos, diminuindo a capacidade dos eritrócitos de navegar na circulação.[29][30] A Hb H também é mais suscetível a lesões oxidativas, provocando precipitação de agregados beta.[26] A oxidação do grupo heme ocasiona a formação de hemicromos que, por sua vez, podem danificar a membrana dos eritrócitos.[31][32] Os danos à membrana dos eritrócitos podem ocasionar o movimento da fosfatidilserina do folheto interno da membrana dos eritrócitos para o folheto externo e a remoção prematura dessas células.[33] Em indivíduos com doença da Hb H, a hemólise pode ser exacerbada pela ingestão de medicamentos oxidativos, infecção e febre. A hemólise também pode ser exacerbada por deficiência de glicose-6-fosfato desidrogenase (G6PD) coexistente, particularmente após exposição a estresse oxidativo. Pacientes com “Hb Constant Spring” (alongamento da globina alfa) e Hb H/Constant Spring também possuem um componente significativo de eritropoiese ineficaz, que contribui para anemia.[28]
Classificação
Tipos e variantes
Há 2 genes da globina alfa em cada cromossomo 16, denominados alfa-2 e alfa-1.[1] Portanto, cada pessoa possui, normalmente, um total de 4 genes funcionais da globina alfa. A talassemia alfa é caracterizada pela menor produção de cadeias de globina alfa secundárias a uma deleção e/ou mutação de ≥1 dos 4 genes da globina alfa.
Classicamente, a talassemia alfa é subdividida em 2 tipos principais: talassemia alfa(0) (--/), em que ambos os genes da globina alfa no mesmo cromossomo sofrem deleção, e talassemia alfa(+) (-alpha/), em que apenas 1 dos 2 genes da globina alfa no cromossomo sofre deleção ou mutação. Grandes deleções envolvendo os genes da globina alfa também podem incluir o elemento regulatório HS-40 ou, raramente, a talassemia alfa(0) pode ser causada por deleções de HS-40 que deixam os genes da globina alfa intactos.[3][4]
As deleções que causam talassemia alfa são mais comuns que as variantes não delecionais. As 7 deleções para talassemia alfa mais comuns são -alfa(3.7) (comum em afro-americanos), -alfa(4.2), (--FIL), (--THAI), (--MED), -(alfa)(20.5), e (--SEA). Em geral, as variantes não delecionais que causam talassemia alfa possuem um fenótipo mais intenso que as variantes delecionais.[5] Aparentemente, isso se deve principalmente a um gene alfa-2 com mutação interferindo na capacidade do gene alfa-1 normal aumentar a produção.[1] Além disso, grandes deleções ou aquelas que afetam o elemento regulatório também podem ocasionar um fenótipo clínico mais intenso.[3]
Classificação genotípica e fenotípica
O fenótipo clínico corresponde ao defeito genético e ao grau de deficiência da síntese da cadeia de globina alfa. O espectro da doença varia de um estado clínico sem nada digno de nota de portador silencioso até a uma talassemia alfa maior, quase sempre fatal na vida uterina se não houver intervenção. A doença da Hb H frequentemente resulta em um fenótipo intermediário, com anemia e hemólise de moderadas a graves.
Existem pelo menos 4 subtipos de talassemia alfa diferentes e distintos.[1]
Portador silencioso de talassemia alfa
O estado de portador silencioso ocorre quando apenas 1 dos 4 genes da globina alfa é afetado.
Os pacientes provavelmente estarão assintomáticos e hematologicamente normais.
A talassemia alfa de portador silencioso também é conhecida como talassemia alfa de traço silencioso, traço de talassemia alfa-2, heterozigosidade para talassemia alfa (+) e talassemia alfa menor.
Traço de talassemia alfa
O traço de talassemia alfa ocorre quando 2 dos 4 genes da globina alfa são afetados: por exemplo, a heterozigosidade para talassemia alfa (0) (ou seja, 2 genes da globina alfa no mesmo cromossomo, em cis, sofrem deleção), ou homozigosidade para talassemia alfa (+) (ou seja, 1 gene da globina alfa em cada cromossomo, em trans, sofre deleção ou mutação).
Os pacientes com traço de talassemia alfa podem apresentar uma leve anemia assintomática e os médicos normalmente diagnosticam esses pacientes, erroneamente, como portadores de anemia ferropriva.
É importante ressaltar que pacientes que são homozigotos para talassemia alfa (+) não delecional podem apresentar manifestações mais graves da doença. Esse é o caso com a Hemoglobina Constant Spring, que é causada por uma mutação no gene da globina alfa-2. Pacientes que são homozigotos para essa mutação (ou seja, ambos os genes da globina alfa-2 são afetados) apresentam um fenótipo clinicamente mais grave que os que são homozigotos para talassemia alfa (+) delecional. Eles apresentam anemia leve com volume corpuscular médio (VCM) normal e hemoglobina corpuscular média (HCM) ligeiramente baixa e frequentemente apresentam icterícia e esplenomegalia.[6]
Doença da hemoglobina H (Hb H)
A doença da hemoglobina H normalmente afeta 3 genes da globina alfa (tetrâmeros beta 4). [Figure caption and citation for the preceding image starts]: Doença da hemoglobina HDo acervo de Elizabeth A. Price e Stanley L. Schrier, Stanford University [Citation ends].
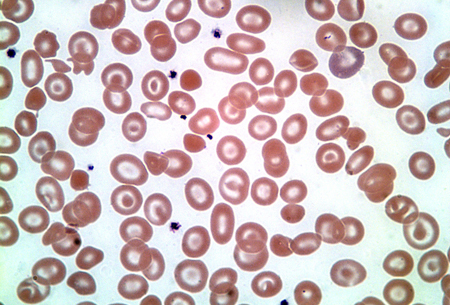 Ela é mais comumente causada pela deleção de 3 genes da globina alfa, mas também pode ser causada pela deleção de 2 genes da globina alfa com mutação pontual de inativação de um terceiro gene.
Ela é mais comumente causada pela deleção de 3 genes da globina alfa, mas também pode ser causada pela deleção de 2 genes da globina alfa com mutação pontual de inativação de um terceiro gene.A doença da Hb H atípica (ou mesmo a hidropisia fetal) também pode ser causada por mutações homozigóticas sem deleção, como mutações no sinal de poliadenilação no gene da globina alfa-2.[7]
A Hb H pode ser detectada no sangue periférico por eletroforese de Hb de rotina. Corpos de inclusão de Hb H também podem ser demonstrados em corante supravital.
Talassemia alfa maior
Normalmente causada quando todos os 4 genes da globina alfa estão deletados.
Ela também é conhecida como síndrome de hidropisia fetal por hemoglobina de Bart ou talassemia alfa (0) homozigótica
O uso deste conteúdo está sujeito ao nosso aviso legal