Abordagem
Há dois aspectos para o diagnóstico: o reconhecimento de um episódio agudo de hipertermia maligna (HM), e a identificação de pacientes com suscetibilidade à HM.
Reconhecimento de um episódio agudo de HM
A HM geralmente ocorre durante o período intraoperatório ou, menos comumente, no pós-operatório. O reconhecimento de um episódio agudo depende de monitoramento intra e pós-operatório adequado:
A ventilação-minuto, o dióxido de carbono exalado (ETCO₂) e as concentrações de oxigênio devem ser documentados. A ventilação-minuto (para manter um dióxido de carbono expirado normal), a produção de dióxido de carbono e o consumo de oxigênio estão aumentados em um evento de HM. Um aumento progressivo inexplicado e inesperado na produção de dióxido de carbono conforme evidenciado no ETCO₂ deve levar a um alto índice de suspeita de HM.[1]
A temperatura central deve ser monitorada durante toda a anestesia. Um aumento acentuado na temperatura central (>40 °C [>104 °F]) pode ser observado na HM.[54][55] Ênfase no monitoramento da temperatura é importante para todos os pacientes aos quais se administra um anestésico, principalmente quando existe a possibilidade de que este cause alterações na temperatura corporal. Foi relatado um aumento nas mortes em decorrência de HM de 2007 a 2012, provavelmente associado a um monitoramento de temperatura inadequado e impreciso.[55]
A menos que a resposta do sistema nervoso simpático seja obnubilada, por exemplo, pelo uso de betabloqueadores ou remifentanil, a produção elevada de dióxido de carbono acompanhada de um aumento inexplicado e inesperado na frequência cardíaca é uma característica diagnóstica de HM. A tendência crescente na frequência cardíaca é mais útil que a obtenção de um valor específico.[1]
O tônus muscular e o grau de bloqueio neuromuscular devem ser verificados. A rigidez muscular do masseter pode ser um sinal precoce de um episódio de HM.[2][6]
Os hábitos e a idade do paciente também deverão ser considerados quando houver suspeita de HM aguda. Uma revisão em pacientes pediátricos revelou diferenças no que diz respeito às características clínicas de episódios de HM aguda entre coortes de idades diferentes na infância.[56] Pacientes mais jovens tem maior probabilidade de apresentar maior grau de acidose láctica, enquanto crianças mais velhas podem apresentar temperatura e níveis de potássio mais altos.
A anestesia inalatória deve ser interrompida, e a taxa de fluxo de gás administrado deve ser aumentado acima da ventilação-minuto se houver suspeita de HM. Adicionar filtros de carvão ativado ao circuito respiratório é uma maneira eficaz de eliminar imediatamente qualquer exposição adicional a agentes voláteis.[57][58] Sangue venoso é obtido para medir os gases e eletrólitos séricos, especialmente potássio. De preferência, a colheita deve ser realizada pelo cateter venoso central se possível. Os achados sugestivos de HM incluem pCO₂ >7.33 kPa (>55 mmHg), pH <7.25, excesso de base mais negativo que -8 mEq/L, e potássio >6 mmol/L (6 mEq/L).[59]
A hipercapnia, a taquicardia e a rigidez muscular podem regredir após a retirada do anestésico inalatório, é realizada uma tentativa terapêutica com dantroleno e o paciente é esfriado até 38ºC (100.4ºF); isso é uma evidência de apoio de que o paciente teve um episódio de HM. No entanto, o dantroleno também pode reduzir o metabolismo, a temperatura, a frequência cardíaca e a acidose em pacientes que não são suscetíveis à HM, então essa resposta não é diagnóstica por si só.
Raramente, a HM pode ser desencadeada sem exposição a anestésicos. Fatores desencadeantes incluem atividade física intensa, rabdomiólise induzida por exercício, doença febril ou episódios repetidos de doença relacionada ao calor.[8][10][11][13] Os pacientes apresentam um episódio espontâneo de rigidez muscular intensa.[7] A HM deve ser considerada no diagnóstico diferencial de qualquer paciente com rigidez muscular associada a qualquer um desses fatores desencadeantes.[3][12]
Exame confirmatório
O teste genético ou teste de contratura muscular pode oferecer um diagnóstico definitivo (veja abaixo). Em ambientes em que a evidência clínica suporta de modo firme um diagnóstico de HM e um teste de contratura muscular é contraindicado (isto é, paciente pesa <20 kg), o teste genético pode preceder o teste de contratura muscular.[60] Nesses pacientes, uma mutação causadora de HM conhecida pode ser encontrada em loci RyR1 (gene do receptor de rianodina), CACNA1S ou STAC3 (uma proteína tubular T específica dos músculos esqueléticos envolvida na regulação de cálcio). Outras mutações diagnósticas estão sendo buscadas.
Exclusão de outros diagnósticos
Como a maioria dos sinais de HM são inespecíficos, é importante descartar outros diagnósticos. Se os resultados da ventilação e de exames de sangue de rotina estiverem normais, a complicação anestésica é improvavelmente decorrente da HM e outras causas deverão ser consideradas. Os diagnósticos mais importantes a serem descartados dependem das anormalidades observadas.
Se a hipercapnia estiver presente, considerar:
Alterações no rendimento do sistema ventilador (vazamentos, falhas nas válvulas)
Mudanças na complacência do sistema respiratório do paciente (vias aéreas superiores comprometidas, pulmões duros, diafragma elevado, retração na direção dos pulmões, rolha de muco)
Administração de medicamentos que aumentam o limiar apneico (narcóticos, sedativos, anestésicos)
Absorção de dióxido de carbono por insuflação
Metabolismo aumentado devido a outras afecções clínicas (sepse, medicamentos estimulantes, supressão de medicamentos, alergia, endocrinopatia).
Se houver febre, considerar:[61]
Superaquecimento iatrogênico
Incapacidade de perder calor devido a roupas oclusivas, alta umidade e temperatura ou vasoconstrição
Doenças que aumentam o ponto de referência da termorregulação
Presença de ingestão recente de drogas ilícitas ou sintéticas, também conhecidas como "novas substâncias psicoativas". Elas podem alterar os níveis de neurotransmissores e resultar em hiperatividade, ocasionando temperaturas elevadas.[62]
Se houver evidência de lesão muscular (incluindo potássio elevado), considerar:
Afecções que produzem isquemia muscular
Possível história de doença muscular hereditária, como distrofinopatia
Medicamentos miopáticos concomitantes como inibidores da HMG-CoA redutase (isto é, estatinas)
Se houver evidência de que a rabdomiólise persiste, isto pode ser uma indicação de defeitos na enzima no músculo (considerar a deficiência de carnitina-palmitoil transferase [CPT] II e deficiência de miofosforilase [doença de McArdle]).
Monitoramento de complicações
Rabdomiólise
Urinálise e mioglobina na urina: devem ser realizadas como um exame inicial em todos pacientes para detectar rabdomiólise. Se houver um achado positivo para sangue na tira reagente, a urina deverá ser enviada para análise microscópica e química para diferenciar entre eritrócitos, mioglobina e hemoglobina.
O débito urinário deve ser monitorado durante o episódio agudo. Uma diminuição no débito urinário acompanhada de mioglobinemia indica uma insuficiência renal iminente.
A creatina quinase deve ser mensurada no momento de um episódio de HM e depois diariamente até que esteja normal. Se a creatina quinase estiver extremamente elevada e houver sinais mínimos de metabolismo aumentado, miopatias estruturais como a distrofinopatia, ou defeitos enzimáticos como a deficiência de CPT, deverão ser considerados como possíveis causas.[37]
Lesão renal aguda
Creatinina sérica: é mensurada para documentar a função renal, que pode estar prejudicada secundariamente à rabdomiólise.
Coagulopatia
Mensurações da função de coagulação como contagem plaquetária, tempo de protrombina, tempo de tromboplastina parcial e fibrinogênio devem ser realizadas como um exame inicial em todos os pacientes com suspeita de ter um episódio de HM. As pessoas susceptíveis à HM podem ter um aumento do risco de sangramento sem exposição aos gatilhos farmacológicos da HM.[63] Considerar a tromboelastografia se possível.
A coagulopatia intravascular disseminada, que causa sangramento excessivo, pode ser uma característica complicadora com a evolução da HM.[1]
Identificação de pacientes com suscetibilidade a HM
A suscetibilidade à HM é uma condição subclínica, apesar de os pacientes poderem descrever desconforto com os exercícios no calor, e alguns se queixarem de cãibras musculares.[12]
Os testes de suscetibilidade à HM por meio de teste de contratura muscular e/ou rastreamento genético fazem parte da manutenção básica da saúde para as famílias afetadas pela HM. Pode haver discordância entre os resultados do teste genético e os do teste de contratura muscular.[7][64] Deve-se confiar nos resultados do teste de contratura muscular para fins de diagnóstico. No entanto, quando uma variante genética causativa é identificada, deve-se aplicar um diagnóstico de suscetibilidade à HM, não sendo necessário o teste de contratura muscular adicional.[60]
[Figure caption and citation for the preceding image starts]: Conduta diagnóstica para a investigação de suscetibilidade à HM. TCIV, teste de contratura in vitro; HM, hipertermia maligna; NSHM (HM negativa ou normal), classificação aplicada quando todos os testes de contratura apresentam resultados negativos; MHShc, MHSh e MHSc, classificações aplicadas quando as respostas de contratura ao halotano e à cafeína são anormais, apenas a resposta ao halotano é anormal ou apenas a resposta à cafeína é anormal, respectivamente. Pacientes com MHShc, MHSh e MHSc devem ser convidados a participar de estudos de pesquisa sobre a base genética da hipertermia malignaHopkins PM et al. Br J Anaesth. 2015 Oct;115(4):531-9. Usado com permissão [Citation ends].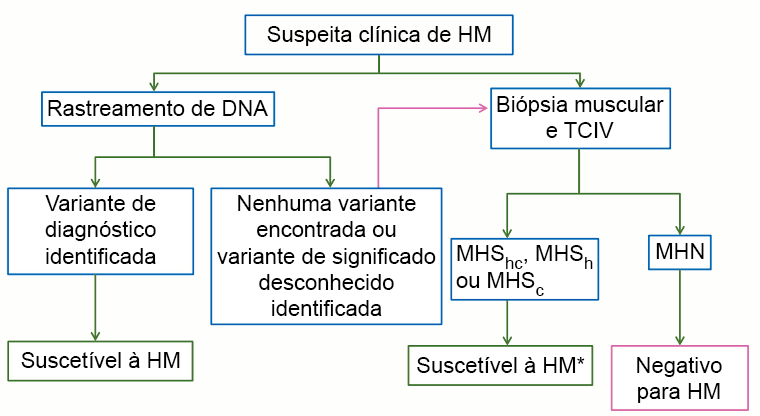
Teste genético
O papel do teste genético na HM é complexo, mas está aumentando.[65][66] Esses testes são realizados por diversas instituições acadêmicas e empresas privadas. A Malignant Hyperthermia Association of the United States (MHAUS), a European Malignant Hyperthermia Group (EMHG) e geneticistas clínicos podem ajudar a identificar as opções de testes genéticos em localidades específicas. Malignant Hyperthermia Association of the United States Opens in new window The European Malignant Hyperthermia Group Opens in new window
Indivíduos com suspeita de terem tido um evento clínico de HM devem ser submetidos a testes genéticos focados nos genes RyR1, CACNA1S e STAC3. Se for descoberto que tais pacientes têm uma variante genética classificada como patogênica ou provavelmente patogênica para HM, eles podem ser considerados suscetíveis à HM sem a necessidade de testes invasivos de contratura. Se nenhuma variante de HM diagnóstica for identificada, o perfil de risco para desenvolver HM sob anestesia permanece incerto, e o teste de contratura deve ser considerado.[60]
De forma separada, há um aumento nos testes genéticos de HM em pacientes sem história pessoal ou familiar de evento anestésico adverso.[67] Como esses pacientes têm uma probabilidade pré-teste substancialmente menor de serem suscetíveis à HM, quaisquer variantes identificadas (especialmente variantes de significância desconhecida) devem ser avaliadas nesse contexto. O aconselhamento genético é indicado em pacientes com mutações suspeitas de causar suscetibilidade à HM, incluindo aquelas de significado desconhecido.[10]
Se houver suspeita da HM como causa de um óbito, deverão ser realizados testes genéticos como parte dos exames post-mortem.[24][65]
Teste de contratura muscular
O teste de contratura muscular é o teste mais sensível para determinar a suscetibilidade à HM. Esse é um teste in vitro que deve ser realizado no músculo fresco, obtido através de biópsias musculares. O paciente precisa ter se recuperado do episódio de HM para o teste ser válido. O paciente deve ir até o laboratório onde o teste muscular será realizado. A biópsia muscular e o teste de contratura devem ser realizados como qualquer cirurgia ambulatorial. A administração in vitro de halotano em bolus, ou o aumento incremental da exposição à cafeína na amostra de músculo fresco, produz respostas mecânicas características à estimulação em músculos suscetíveis à HM, que são diagnósticas.[68][Figure caption and citation for the preceding image starts]: Resposta mecânica do músculo normal e do músculo suscetível à hipertermia maligna (hipertermia maligna [HM]; positivo) à estimulação direta na presença de uma dose de halotano a 3%Do acervo da Dra. Sheila Muldoon [Citation ends].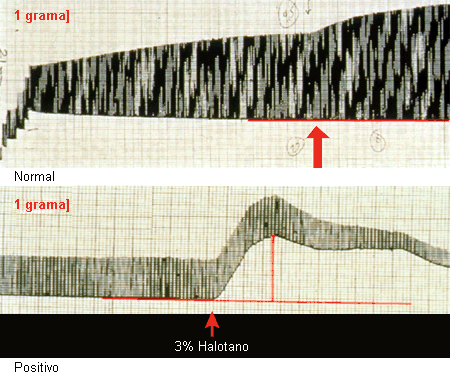 [Figure caption and citation for the preceding image starts]: Resposta mecânica do músculo normal e do músculo suscetível à hipertermia maligna (hipertermia maligna [HM]; positivo) à estimulação direta na presença da administração incremental de cafeínaDo acervo da Dra. Sheila Muldoon [Citation ends].
[Figure caption and citation for the preceding image starts]: Resposta mecânica do músculo normal e do músculo suscetível à hipertermia maligna (hipertermia maligna [HM]; positivo) à estimulação direta na presença da administração incremental de cafeínaDo acervo da Dra. Sheila Muldoon [Citation ends].
O teste de contratura a cafeína-halotano (TCCH) e o teste de contratura in vitro (TCIV) são os bioensaios padronizados utilizados.[69] Esses testes são desenvolvidos para serem bem sensíveis e são os únicos testes que podem definitivamente descartar o diagnóstico de HM ou suscetibilidade à HM. A resposta característica é quantificada como um aumento no limiar de contratura. Os critérios de diagnóstico diferem dependendo de qual teste é usado:
TCCH: os pacientes com um limiar de contratura aumentado em resposta à cafeína ou halotano são considerados suscetíveis à HM.
TCIV: os pacientes com um aumento do limiar de contratura em resposta à cafeína e ao halotano, ou apenas a um desses agentes, são diagnosticados como sendo suscetíveis à HM.[60]
Os valores preditivos positivo e negativo de ambos os testes de contratura dependem da probabilidade prévia de o paciente ser suscetível à HM, o que é estabelecido por meio de uma análise minuciosa da história médica e anestésica pessoal e familiar.[7]
Esses testes necessitam de uma cirurgia pequena e devem ser realizados em um centro de diagnóstico de HM, os quais são vários ao redor do mundo. The European Malignant Hyperthermia Group Opens in new window Malignant Hyperthermia Association of the United States Opens in new window Esses testes não foram padronizados para a população pediátrica e como eles requerem um grande pedaço de músculo eles não são adequados em crianças <20 kg.
Rastreamento de parentes
Se um paciente é diagnosticado como suscetível à HM, os familiares consanguíneos devem ser rastreados assim que a avaliação do paciente estiver completa.[24] Se o paciente apresentar uma mutação identificável, estudos genéticos em parentes podem focar em um éxon em particular.
Se a mesma mutação foi identificada em um parente, este indivíduo deve ser considerado suscetível à HM, e o teste de contratura muscular não é necessário. Se uma mutação não for identificada, o teste de contratura muscular será necessário para descartar definitivamente o diagnóstico.[70]
Muitas famílias apresentam mutações novas no RyR1.[11][13][15][18][19][25][26][71] Suspeita-se que isso cause HM, mas não há confirmação biológica.[34][72] Dada a penetrância variável e a prevalência de indivíduos suscetíveis à hipertermia maligna (SHM), novas técnicas genéticas como o sequenciamento do exoma identificaram famílias não suspeitas como SHM.[20][33][65]
Detecção de deficiências de enzimas musculares
A HM é comumente associada com um aumento na creatina quinase, e alguns pacientes com suscetibilidade à HM apresentarão elevações crônicas. Se um paciente apresenta uma elevação recorrente da creatina quinase >5 vezes o limite superior do normal, as deficiências enzimáticas musculares associadas à rabdomiólise devem ser investigadas.[37][51] O perfil de intolerância ao exercício envolve testes específicos para essas deficiências, incluindo deficiência de carnitina-palmitoil transferase 2 (CPT2), deficiência de miofosforilase e deficiência de mioadenilato deaminase.
O uso deste conteúdo está sujeito ao nosso aviso legal