Etiologia
A etiologia da doença de Kawasaki (DK) permanece desconhecida. No entanto, as seguintes observações sugerem que a doença é desencadeada por um agente desconhecido em um hospedeiro suscetível.
Variação sazonal: variação na incidência associada com estações diferentes; observam-se picos no inverno e na primavera no Japão e nos EUA.[11]
Distribuição geográfica: foram relatadas agrupações temporais nos EUA, no Japão e ao redor do mundo.[3] Além disso, foram observados no Japão pontos de aumento da incidência em determinadas áreas geográficas.[14]
Idade de início: o pico da incidência ocorre na primeira infância, com 80% dos casos em crianças <5 anos. A raridade de casos em lactentes <3 meses de idade pode sugerir anticorpos protetores transplacentários.[3][9]
Até o momento, nenhum fator desencadeante infeccioso foi identificado para a DK; especificamente, os estudos não encontraram nenhuma relação com parvovírus B19, retrovírus, vírus Epstein-Barr, herpes, sarampo ou coronavírus humano (NL-63).[15]
Fenótipos similares de doenças mediadas por toxinas (inclusive síndrome do choque tóxico estreptocócico e estafilocócico e escarlatina) com febre, comprometimento da membrana mucosa e erupção cutânea com descamação também fizeram com que determinadas bactérias fossem consideradas fatores desencadeantes para DK. Além disso, há relatos de que o Staphylococcus aureus que secreta a toxina da síndrome do choque tóxico, isolado de um paciente com DK, se manifestou com aneurisma coronariano.[16] Especula-se que um fator desencadeante, potencialmente uma infecção, resulta em uma reação imunologicamente mediada, que causa a manifestação da doença em um hospedeiro imunogeneticamente suscetível. Foi proposto que, tanto na DK quanto na SCT, a doença é causada por toxinas virais e bacterianas atuando como superantígenos.[17][18] A teoria de superantígeno para a DK continua a ser investigada; entretanto, não há dados significativos para dar suporte no momento.[19]
A genética da DK é complexa, e é provável que múltiplos polimorfismos contribuam para a patogênese. Acredita-se que a genética desempenhe um papel importante, dada a incidência elevada da DK em populações asiáticas nos EUA e em outras áreas geográficas. Além disso, há uma agregação familiar, com aumento da incidência em pais e irmãos da criança afetada.[11] No maior estudo de associação genômica ampla da DK no Japão, determinou-se que as suscetibilidades genéticas associadas de forma mais significativa foram de variantes com alta afinidade do receptor Fc para a imunoglobulina G (gene FCGR2A) e de variantes relacionadas ao regulador da região do receptor de células T, conhecido como inositol 1,4,5-trifosfato 3-quinase C (ITPKC).[11][20] As variantes deste último estão associadas com o desenvolvimento de aneurismas de artéria coronária e com a resistência à imunoglobulina intravenosa.[11]
Uma metanálise de estudos de associação genética, realizada em 2017, constatou que 23 polimorfismos de genes estão associados com a suscetibilidade à DK, e que 10 podem estar associados com lesões em artérias coronárias. Muitos desses genes são ativadores ou inibidores de linfócitos; outros estão envolvidos na função das citocinas, quimiocinas e moléculas de adesão, ou no remodelamento vascular.[21]
Embora existam semelhanças entre DK e acrodinia (hipersensibilidade ao mercúrio), os estudos que relacionaram a DK a medicamentos, toxinas, produtos químicos e metais pesados mostraram resultados negativos.[22]
Fisiopatologia
Inicialmente, a DK foi considerada uma doença benigna autolimitada; no entanto, sabe-se agora que 20% a 25% dos pacientes não tratados desenvolvem aneurismas de artéria coronária, e há uma taxa de mortalidade de até 2% associada com os aneurismas de artérias coronárias. Além disso, relatou-se que a DK está associada com infarto do miocárdio, morte súbita e cardiopatia isquêmica.[23]
Na fase precoce da doença, há o desenvolvimento de edema e a infiltração de neutrófilos na parede da artéria coronária, com uma transição rápida para células mononucleares.[24] Isso é seguido pela produção local de metaloproteinases da matriz que causam destruição da parede elástica interna e da camada média. A proliferação miofibroblástica íntima causa substituição do tecido conjuntivo fibroso da íntima e da média, levando à formação de aneurismas, fibrose e estenose.[25] Foram identificadas alterações fibróticas no miocárdio, enfatizando que a inflamação cardíaca pode ser mais disseminada do que foi considerado inicialmente, e não restrita às artérias coronárias.[12]
O remodelamento arterial, um processo que pode ocorrer ao longo de anos, pode causar estenose, particularmente na interface entre a artéria normal e o aneurisma. Os aneurismas maiores têm maior probabilidade de persistir, e estão associados com o aumento do risco de oclusão coronariana trombótica, estenoses e infarto do miocárdio.
Há relatórios disponíveis de crianças com DK que não desenvolveram anomalias coronárias durante a fase aguda da doença e morreram anos depois devido a causas não relacionadas. As autópsias realizadas nessas crianças demonstraram espessamento da camada íntima da artéria coronária e fibrose na média.[26] Não se sabe ao certo se esses casos representam sequelas tardias de aneurismas ausentes após a apresentação aguda; ou, o que é mais preocupante, o surgimento de vasculopatia de artérias coronárias por DK tardia, independentemente da presença de aneurismas de artérias coronárias após a doença aguda.
Classificação
Estágios clínicos[1]
Clinicamente, a evolução da doença de Kawasaki (DK) não tratada é dividida nos seguintes estágios:
Estágio febril agudo (com duração de 1-2 semanas)
Febre, irritabilidade, adenite cervical, conjuntivite, erupção cutânea, eritema da mucosa, eritema doloroso das mãos e dos pés, artralgia ou artrite, possível miocardite e pericardite.
Estágio subagudo (com duração de 2-4 semanas)
Febre, erupção cutânea e linfadenopatia foram revertidas; se a febre persistir, há aumento do risco de complicações cardíacas; irritabilidade persistente, inapetência e hiperemia conjuntival; a descamação dos membros começa nesta fase.
O paciente pode estar completamente assintomático se a imunoglobulina intravenosa (IGIV) for administrada. A descamação periungueal pode ser a única manifestação clínica aparente.
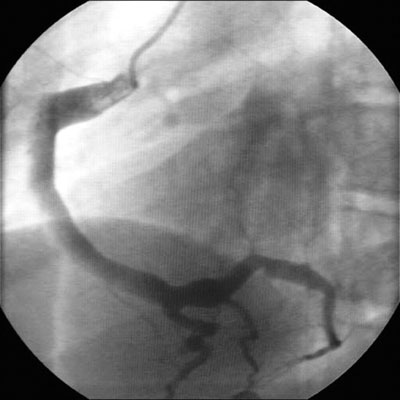
As anormalidades cardíacas (ectasia da artéria coronária ou aneurismas) podem se desenvolver durante esta fase e, raramente, depois nos pacientes tratados com IGIV.[Figure caption and citation for the preceding image starts]: Ectasia da artéria coronáriaBMJ Case Reports 2009; doi:10.1136/bcr.10.2008.1113 [Citation ends].
Convalescência (com duração de 4-8 semanas)
Todos os sinais de inflamação desapareceram, e os marcadores da fase aguda estão normalizados.
Se presentes, ectasia da artéria coronária ou aneurismas podem persistir e aumentar.
Estágio crônico (variável)
Se presente, a dilatação da artéria coronária pode ter sido revertida.
No entanto, os aneurismas da artéria coronária podem persistir pela vida adulta. Esses pacientes correm o risco de trombose da artéria coronária subsequente, ruptura e infarto do miocárdio.
O uso deste conteúdo está sujeito ao nosso aviso legal