Abordagem
A esferocitose hereditária (EH) é mais comumente diagnosticada como uma doença leve em crianças, na qual pode haver icterícia (gravidade variável com o tempo) e esplenomegalia. Exames laboratoriais de rotina geralmente confirmam o diagnóstico pela detecção de:
Aumento da renovação de células eritrocitárias (reticulocitose) ± anemia
Esferócitos típicos no esfregaço sanguíneo
Ausência de uma causa imune associada (teste de antiglobulina direto negativo [TAD]).
Exames laboratoriais confirmatórios podem ser necessários em pessoas com características atípicas.[19] Testes especializados adicionais são raramente necessários.
Quando se suspeita de um diagnóstico de EH, os pacientes devem ser encaminhados a um hematologista para diagnóstico definitivo e acompanhamento de rotina.
[Figure caption and citation for the preceding image starts]: Algoritmo de diagnóstico para esferocitose hereditária (EH)Copyright © BMJ Best Practice - Todos os direitos reservados [Citation ends].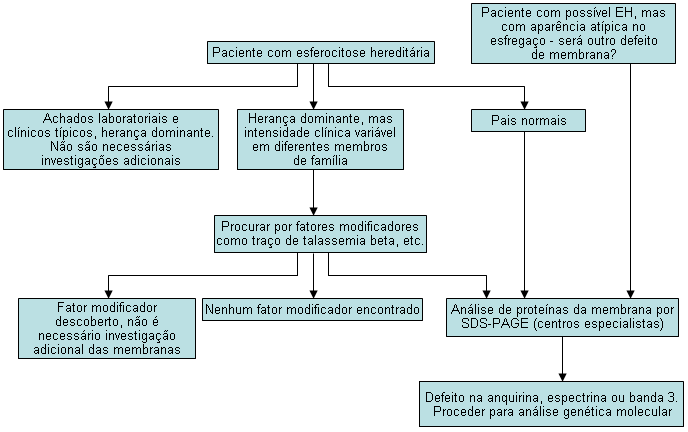
História
A EH pode estar presente em todas as populações, mas é mais comum em descendentes de pessoas do norte da Europa.[14] Entretanto, a EH é rara em indivíduos negros.[17] A maioria das pessoas com EH apresentarão história familiar de anemia, icterícia, esplenectomia ou EH conhecida.
A descoberta pode ocorrer em qualquer idade. Indivíduos afetados mais gravemente tendem a apresentar a doença mais precocemente. A gravidade clínica varia de ausência de sintomas a fadiga decorrente de anemia grave e icterícia.[4] Frequentemente, quando não há sintomas, a doença é detectada como um achado incidental quando um exame de sangue é realizado por outro motivo.
A infecção por parvovírus B19 pode revelar uma EH não diagnosticada
Crianças e adultos podem ser assintomáticos até que contraiam infecção pelo parvovírus B19 resultando em uma crise aplástica.[20][21] O parvovírus B19 ataca diretamente os precursores eritroides na medula óssea resultando em aplasia eritrocitária por cerca de 10 dias. Pacientes com uma meia-vida eritrocitária reduzida, como ocorre na EH (e em outras doenças hemolíticas), têm uma anemia rapidamente progressiva durante esse período de eritropoiese ausente, e tipicamente apresentam início agudo de palidez, letargia e febre acentuadas. O valor da hemoglobina (Hb) é geralmente entre 3 e 6 g/dL. A contagem de reticulócitos é <1%.
Durante a fase de recuperação, a contagem de reticulócitos aumenta, e a imunidade ao parvovírus B19 geralmente ocorre após a recuperação de uma crise aplástica.
Os sintomas e sinais de EH podem variar com o tempo
Crises hiper-hemolíticas são mais comuns, mas menos graves que os episódios aplásticos, e são caracterizadas pela aceleração do processo hemolítico normal que resulta em exacerbação dos sintomas. Esses episódios geralmente acompanham infecções virais inespecíficas e podem ser repetitivos.
Em contraste à crise aplástica, a hemoglobina (Hb) está geralmente no intervalo de 50 a 80 g/L (5-8 g/dL) (ou até mais alta durante episódios mais leves), a contagem de reticulócitos está elevada, e o paciente apresenta-se mais ictérico que o normal. Pode haver uma esplenomegalia aguda e significativa com dor abdominal no quadrante superior esquerdo e sintomas de saciedade precoce. O baço geralmente regride para o tamanho prévio entre episódios.[Figure caption and citation for the preceding image starts]: Apresentação da esferocitose hereditária (EH) por idadeCopyright © BMJ Best Practice - Todos os direitos reservados [Citation ends].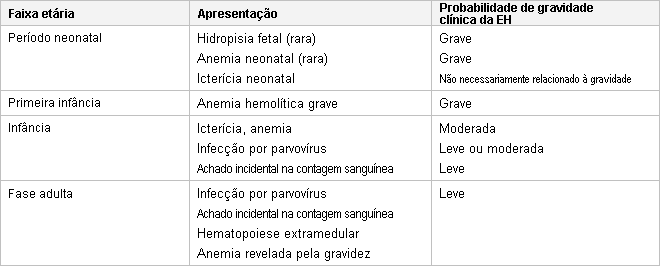
Exame físico
Muitos indivíduos com EH apresentam uma icterícia leve. É uma característica comum quando a EH se apresenta no período neonatal.[4] A gravidade da icterícia não está necessariamente relacionada a uma gravidade futura da EH.
Em alguns casos, a icterícia é grave o suficiente para justificar uma exsanguineotransfusão.[7] Muito raramente, a EH pode apresentar hidropisia fetal ou fetos natimortos devido à anemia grave.[4][22]
O diagnóstico de EH deve ser considerado em qualquer indivíduo com esplenomegalia inexplicável ou inesperada em qualquer idade. A esplenomegalia (aumento leve a moderado) é muito comum em todos os níveis de gravidade de EH, mas não é específica desse distúrbio.[7] A esplenomegalia normalmente não apresenta quaisquer sintomas ou consequências clínicas. No entanto, durante crises hiper-hemolíticas, o baço raramente pode estar aguda e significativamente aumentado, e causar dor abdominal no quadrante superior esquerdo e sintomas de saciedade precoce. O baço geralmente regride para o tamanho prévio entre episódios.
Indivíduos com EH podem ou não apresentar sinais de anemia.[4] Dependendo da gravidade da doença, os sinais de anemia serão variáveis: de nenhum sinal óbvio a palidez intensa.[Figure caption and citation for the preceding image starts]: Esclera ictérica no olho de uma criança com esferocitose hereditária (EH)Do acervo de Paula Bolton-Maggs, University of Manchester, Reino Unido; usado com permissão [Citation ends].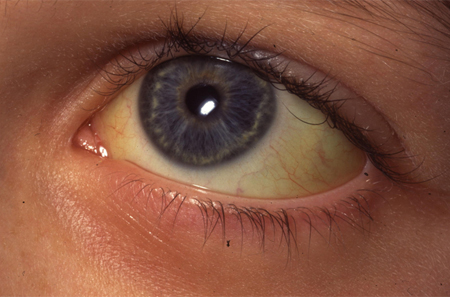
Exames iniciais
Muitos casos podem ser detectados como um achado incidental quando um hemograma completo é realizado por outro motivo. O diagnóstico também deve ser considerado em qualquer paciente com icterícia ou esplenomegalia, ou história familiar de EH em um parente de primeiro grau.[23]
O diagnóstico deve ser considerado na presença de um resultado de Hb baixo e inesperado em qualquer idade.
Hemograma completo e esfregaço
Em muitos casos, o diagnóstico é feito por contagem e esfregaço sanguíneos. O paciente tem uma hemoglobina normal ou reduzida, VCM normal ou reduzido e, frequentemente, um CHCM elevado (>350 g/L [>35 g/dL]).[4][23] A morfologia eritrocitária anormal dos esferócitos está presente no esfregaço sanguíneo, com reticulócitos aumentados.[4] O esfregaço sanguíneo pode revelar eritrócitos pinçados (em forma de cogumelo) em associação com esferócitos, que são decorrentes de mutações das proteínas banda 3.[23] A anemia pode estar ausente na EH quando a capacidade de produção da medula aumenta o suficiente para manter a hemoglobina (Hb) normal, mas haverá um aumento nos reticulócitos (conhecido como hemólise compensada).
O esfregaço sanguíneo neonatal pode ser difícil de interpretar. Uma análise cuidadosa do esfregaço sanguíneo é necessária com acompanhamento se o diagnóstico não estiver claro. Apresentações típicas de esferócitos no esfregaço sanguíneo são mais fáceis de observar após alguns meses.
Diagnósticos diferenciais
Uma análise cuidadosa da morfologia dos eritrócitos do esfregaço sanguíneo é muito importante para não deixar de considerar distúrbios alternativos menos comuns.[24] A presença de esferócitos não está limitada à EH, e outros diagnósticos devem ser excluídos através de história, contexto clínico e investigações adequadas.[24]
O diferencial mais importante é a anemia hemolítica autoimune (AHAI). Na AHAI, anticorpos anormais cobrem os eritrócitos e podem ser detectados pelo TAD, que é negativo na EH. Na primeira infância, a hemólise causada por anticorpos imunoglobulina G (IgG) maternais irregulares também deve ser excluída como uma possível causa.
É importante diferenciar a EH de outros distúrbios hereditários da membrana eritrocitária. Muitos desses distúrbios são óbvios em apresentações do esfregaço sanguíneo (por exemplo, eliptocitose), mas quando as apresentações eritrocitárias são atípicas, outros diagnósticos devem ser considerados. Isso é particularmente importante para descartar estomatocitose hereditária com defeitos de transporte da membrana, pois a esplenectomia possui um alto risco de tromboembolismo venoso pós-operatório.[25] Exames de bilirrubina sérica e aminotransferases hepáticas são realizados se houver suspeita clínica de icterícia. Casos de anemia diseritropoética congênita foram diagnosticados erroneamente como EH.[26][27][Figure caption and citation for the preceding image starts]: Esfregaço sanguíneo de um paciente com esferocitose hereditária (EH); esferócito indicadoDo acervo de Shelley Crary, University of Texas Southwestern Medical Center, TX; usado com permissão [Citation ends].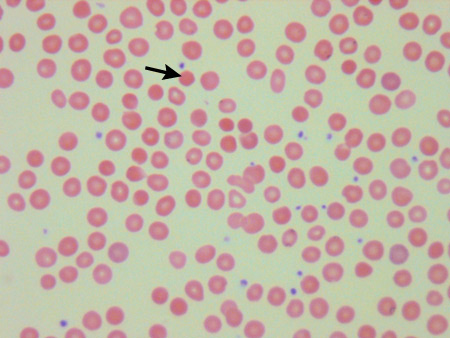 [Figure caption and citation for the preceding image starts]: Esfregaço sanguíneo de paciente com esferocitose hereditária (A) comparado ao sangue normal (B); Eritrócito pinçado (em forma de cogumelo) indicadoDo acervo de Paula Bolton-Maggs, University of Manchester, Reino Unido; usado com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Esfregaço sanguíneo de paciente com esferocitose hereditária (A) comparado ao sangue normal (B); Eritrócito pinçado (em forma de cogumelo) indicadoDo acervo de Paula Bolton-Maggs, University of Manchester, Reino Unido; usado com permissão [Citation ends].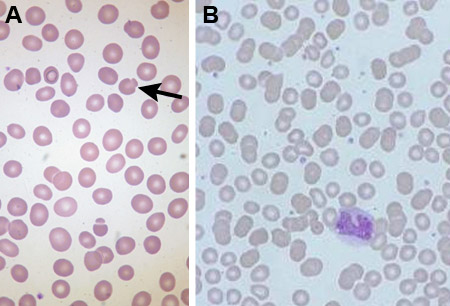
Exames confirmatórios a serem considerados
O caso típico de EH não requer nenhum exame confirmatório. Se o diagnóstico for ambíguo, um teste confirmatório pode ser considerado.
Teste de ligação eosina-5-maleimida (EMA)
A confirmação por meio do teste de ligação da eosina-5-maleimida é recomendada quando as características não são típicas (por exemplo, a morfologia no esfregaço sanguíneo não é muito típica ou não há história familiar).[19] O teste de ligação EMA baseia-se na intensidade de fluorescência analisada por citometria de fluxo.[28][29][30]
O EMA liga-se à proteína banda 3, que está rompida na EH, resultando em uma fluorescência diminuída. O teste de ligação da EMA foi relatado como tendo uma sensibilidade de 92.7% para EH e uma especificidade de 99.1%, mas pode estar anormal em outros distúrbios eritrocitários (particularmente em anemia diseritropoética congênita do tipo II).[28]
Teste de lise pelo glicerol acidificado
Utiliza 20 microlitros de sangue total e mede o tempo necessário para que a absorbância caia para metade do valor original usando uma suspensão eritrocitária antes e depois da adição de glicerol. Esse teste revelou uma sensibilidade de 98.3% para EH e uma especificidade de 91.1%, mas também é positivo para AHAI, gravidez, mielodisplasia e algumas outras condições.[31]
Teste de crio-hemólise
Eritrócitos com defeitos na membrana são suscetíveis a lise quando sujeitos a frio intenso. A sensibilidade e a especificidade de 95% e 96%, respectivamente, foram relatadas para EH usando um limiar de crio-hemólise de 15%.[32]
Análise genética
Usado em associação com testes padrão.[4] Mutações foram descritas em cinco proteínas citoesqueléticas (alfa e beta-espectrina, anquirina, banda 3 e proteína 4.2).[6][33]
Mutações nos genes para anquirina, banda 3 ou beta-espectrina podem causar um defeito secundário em outras proteínas esqueléticas. A EH não dominante pode ocorrer como resultado da herança de uma mutação patogênica de um dos pais e de um alelo pouco expressivo silencioso do outro, ou de mutações de novo. Mutações no gene da proteína 4.2 são mais comuns entre pacientes japoneses com EH não dominante.[34][35]
Exames adicionais em casos difíceis
O caso típico de EH não requer exames adicionais. Exames adicionais são indicados:
Quando o fenótipo clínico é mais grave que o esperado pelas aparências eritrocitárias
Quando as anormalidades eritrocitárias são mais graves que as observadas em um dos pais afetados
Quando a esplenectomia é considerada e a morfologia é atípica.
A análise proteica quantitativa por eletroforese em gel de poliacrilamida contendo dodecil sulfato de sódio (SDS-PAGE) pode ser realizada em casos atípicos. A SDS-PAGE analisa o conteúdo da membrana eritrocitária e estabelece qual proteína de membrana é deficiente por meio da demonstração de bandas de espectrina, anquirina, banda 3 e proteína 4.2 (as proteínas de membrana que podem ser afetadas na EH).[5][19][36][37][38][39]
É importante descartar formas raras de distúrbios eritrocitários onde a esplenectomia é contraindicada.[7] A esplenectomia tem pouco valor na anemia diseritropoética congênita tipo II, que pode ser confundida com EH.
O uso deste conteúdo está sujeito ao nosso aviso legal