Abordaje
La miastenia grave (MG) afecta con más frecuencia a las mujeres adultas jóvenes (menores de 40 años) y a los hombres de edad avanzada (mayores de 60 años), pero puede ocurrir a cualquier edad.[36][37]
Tras una revisión de la historia clínica del paciente y los hallazgos típicos de la exploración, se puede establecer el diagnóstico de MG mediante pruebas clínicas y serológicas. Si las pruebas serológicas no aportan hallazgos relevantes, se deben realizar pruebas clínicas neurofisiológicas. Las pruebas de función pulmonar pueden ayudar a pronosticar si la respiración está alterada, y así ayudar a evitar la crisis miasténica.
Antecedentes
Las características típicas son debilidad y fatiga de los músculos esqueléticos con una distribución característica. La enfermedad suele presentarse con una de las tres formas diferentes: ocular, orofaríngea o generalizada.
Según la forma inicial predominante, los pacientes con MG pueden referir una multitud de síntomas que incluyen ptosis, diplopia, disartria (trastorno del habla), disfagia (dificultad con la deglución), paresia facial, debilidad en las extremidades proximales y disnea. Es característico que la debilidad de las extremidades empeore con la actividad (fatiga) y mejore con el reposo, y las fluctuaciones a menudo (pero no siempre) muestran una variación diurna (mejor por la mañana que por la tarde).
Con menor frecuencia, los pacientes pueden presentar una debilidad predominante o exclusiva de las extremidades, lo que a veces se denomina MG de la cintura.
Exploración física
Generalmente la gravedad clínica de la miastenia grave (MG) se clasifica según la función y la región.[4][68][69]
La debilidad muscular fatigable puede ser localizada o generalizada, e involucra los párpados, los músculos extraoculares del ojo, el rostro, la orofaringe, el cuello, los músculos respiratorios y las extremidades. La ptosis y la diplopia se presentan de manera temprana en la mayoría de los pacientes.[22] Para comprobar el tiempo de la ptosis (que por lo general, es de más de 3 minutos en personas sanas) se solicita al paciente que mire hacia arriba y se mide el lapso de tiempo hasta la aparición de la ptosis. Levantar el párpado superior puede inducir una ptosis en el párpado contralateral. El enfriamiento del párpado durante 2 a 5 minutos con una bolsa de hielo mejora la ptosis en más del 95% de los pacientes con MG, pero puede no mejorar la ptosis grave.[70][71]
Entre el 50% y el 60% de los pacientes que presentan síntomas puramente oculares evolucionarán hasta desarrollar una MG generalizada, y la gran mayoría lo hará en los primeros 1 ó 2 años.[23][Figure caption and citation for the preceding image starts]: Ptosis del párpado superior izquierdo en un paciente con miastenia graveForoozan R, Sambursky R. Ocular myasthenia gravis and inflammatory bowel disease: a case report and literature review. Br J Ophthalmol. 2003 Sep;87(9):1186-7. [Citation ends].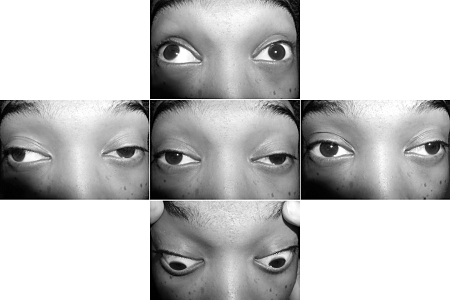
Cuando se ven afectados los músculos faciales y orofaríngeos, puede presentarse sonrisa aplanada o transversa característica o voz nasal y dificultad para masticar y deglutir.
Una exploración física de las extremidades muestra debilidad de los músculos proximales con fatiga. El tiempo de abducción de los brazos se comprueba pidiendo a los pacientes que mantengan los brazos extendidos; las personas sanas suelen poder mantener los brazos en alto durante más de 3 minutos.
No debe haber un desgaste muscular evidente (a excepción de la lengua y, con menor frecuencia, del paladar en los pacientes con MG con anticuerpos contra la tirosina cinasa específica del músculo [MuSK]), y los reflejos son normales. Las sensaciones están intactas (a menos que haya otro trastorno) y no hay disfunción autonómica.
En los casos de crisis miasténica existe afectación respiratoria grave y/o afectación bulbar.
Características de la MG con MuSK
Los pacientes con MG con MuSK presentan uno de los tres fenotipos; los dos primeros se consideran característicos:[5][24]
Debilidad facio-faríngea grave, con atrofia de los músculos implicados en la enfermedad de larga duración
Debilidad predominante en el cuello y en las vías respiratorias, con avance frecuente hacia una crisis miasténica, o
Las características clínicas son indistinguibles de las de la MG no MuSK.
Las siguientes características clínicas distinguen la MG con MuSK positiva de la MG con MuSK negativa:[5]
Las características oculares no son comunes pero pueden presentarse.
La atrofia facial y la atrofia de la lengua se determinan mediante exploración física y resonancia magnética (IRM).
La debilidad en las extremidades es leve.
Los pacientes con debilidad muscular prominente en el cuello y la respiración pueden presentar pruebas de electrodiagnóstico normales en los músculos de la cara y las extremidades. Es necesario realizar pruebas en los músculos implicados para establecer el diagnóstico.
La debilidad tiende a ser más grave; el 23% de los pacientes evolucionaron hacia una crisis y el 85% presentaban una clase máxima de Miastenia Gravis Foundation of America (MGFA) de III o superior en una clínica.
Respuesta clínica deficiente al tratamiento con inhibidores de la colinesterasa.
Características de LRP4, cortactina, agrina y colágeno Q MG
Se ha informado que los pacientes con MG y anticuerpos contra el LRP4 suelen tener una MG leve a moderada, similar a la de algunos pacientes con anticuerpos contra el receptor de acetilcolina (AChR); en un estudio se informó de todo el espectro de gravedad de los síntomas.[9][10][13][50][72] Se necesitan estudios adicionales para caracterizar completamente los fenotipos clínicos, así como cualquier patrón de edad de inicio, diferencias de sexo y grupos étnicos/raciales, para la LRP4.
Se ha encontrado que un pequeño número de pacientes con anticuerpos contra la cortactina y sin anticuerpos detectables contra el AChR (utilizando las pruebas estándar) o la MuSK presentan una enfermedad leve a moderada, incluida la MG ocular, similar al patrón observado en la MG por AChR.[17][18][19][21] Se desconoce si alguno de estos pacientes presentaba anticuerpos contra el LRP4, la agrina o el colágeno Q, o anticuerpos contra el AChR que solo se detectarían con la tecnología de AChR agrupado basada en células.
Aún no se han establecido las características de la MG en pacientes con anticuerpos contra la agrina o el colágeno Q.
Visión general de las investigaciones
La MG es un diagnóstico clínico apoyado por pruebas serológicas, electrofisiológicas y farmacológicas.
Pruebas serológicas
Todos los pacientes deben someterse a pruebas serológicas.
Ensayo de unión y modulación de anticuerpos contra el AChR: los anticuerpos contra el AChR se detectan en el 80% al 90% de los pacientes con MG generalizada y hasta el 50% de los pacientes con MG ocular, con una especificidad del 99% en ambos casos.[70] El título absoluto no se correlaciona con la gravedad de la MG, aunque en algunos estudios se ha sugerido que los niveles altos de anticuerpos moduladores del AChR se correlacionan con la gravedad.[2][22]
Ensayo basado en células de anticuerpos AChR: identificó pacientes con MG que, en general, parecían similares a los pacientes identificados por las pruebas estándar de AChR, con un mayor porcentaje de enfermedad de inicio prepuberal.[73]
Prueba de anticuerpos MuSK: si la prueba estándar de anticuerpos AChR es negativa, procede a la prueba serológica de anticuerpos MuSK. Los anticuerpos MuSK se detectan hasta en el 70% de los pacientes con MG generalizada que son seronegativos para los anticuerpos AChR.[5][62] En los Estados Unidos, los anticuerpos MuSK son frecuentes en la población de raza negra; el 70% de las mujeres de raza negra con MG seronegativa generalizada tienen anticuerpos MuSK.[5]
Pruebas de anticuerpos frente al receptor estriado: las pruebas de anticuerpos contra la titina y la rianodina no se realizan de forma rutinaria en todos los pacientes, y los anticuerpos son raros en los pacientes sin timoma, aparte de los pacientes de "inicio de mayor".[74] Se detectan en el 75% al 95% de los pacientes con timoma y MG, y su presencia puede indicar recurrencia del timoma.[75]
Las pruebas de anticuerpos contra el LRP4, la agrina, el colágeno Q o la cortactina no están ampliamente disponibles, y no está claro hasta qué punto son específicos para la MG, pero los anticuerpos contra el LRP4 pueden resultar útiles en el entorno clínico adecuado.[10] Los anticuerpos contra la cortactina se observan en las miopatías, lo que sugiere que el análisis de los anticuerpos contra la cortactina no sería útil como prueba de diagnóstico de la MG.[17][19]
Pruebas electrofisiológicas
Si las pruebas serológicas no son destacables o hay rasgos clínicos atípicos, deben realizarse pruebas electrofisiológicas.
La estimulación nerviosa repetitiva (ENR) a velocidad lenta debería revelar una disminución. En este procedimiento, se envían choques eléctricos al nervio y se registran los potenciales de acción mediante electrodos de superficie colocados sobre el músculo.[70] Una mejoría en decremento de la línea de base tras ejercicios breves (facilitación postejercicio) y un decremento acentuado tras 2 a 3 minutos de postejercicio (agotamiento postejercicio) son características típicas del electrodiagnóstico. La sensibilidad es del 79% en la MG generalizada y del 50% en la MG ocular, con una especificidad del 97%.[70] Se ha sugerido una disminución del 7% al 8% como límite para los músculos faciales.[76] Si se aumenta el número de músculos examinados, aumentará la probabilidad de demostrar un decremento significativo.[77]
Si la estimulación nerviosa repetitiva (ENR) es negativa o equívoca, se recomienda el EMG de fibra única (SFEMG). El SFEMG mide la transmisión de la unión neuromuscular (UNM) entre 2 o más fibras musculares adyacentes inervadas por el mismo axón motor. El SFEMG puede mostrar un aumento de la variabilidad en las latencias motoras (fluctuación) o una insuficiencia total de la transmisión de la UNM (bloqueo) en las fibras musculares. Cuando se realiza en los músculos faciales, la prueba es positiva en el 86% al 92% de los pacientes con MG ocular, con una especificidad del 70% al 96%. En los pacientes con MG generalizada, la especificidad y la sensibilidad son de hasta el 98%.[70] Esta prueba no es específica para la MG, y es importante estar seguro de que no hay evidencia de denervación o miopatía en los músculos examinados.
Estudios por imágenes
Se debe realizar una tomografía computarizada (TC) del tórax en todos los pacientes recién diagnosticados para detectar timoma (que ocurre en aproximadamente el 15% de los pacientes con miastenia grave [MG]) o hiperplasia del timo (que ocurre en el 75% de los pacientes con MG).[Figure caption and citation for the preceding image starts]: Tomografía computarizada que muestra una masa mediastínica anteriorHamid UI, Jones JM. Masa mediastínica. BMJ Case Reports 2010 Oct 4;2010:bcr1120092471. [Citation ends].
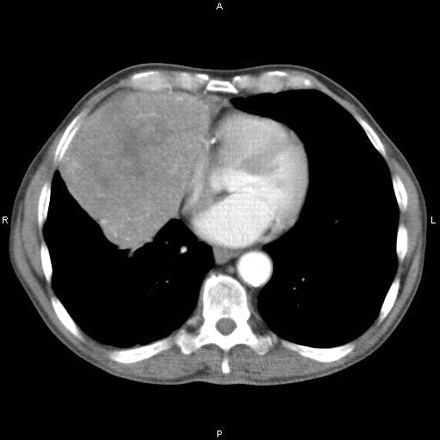
La resonancia magnética de rutina no ofrece ningún beneficio adicional. La resonancia magnética con desplazamiento químico y ponderación de la difusión puede identificar la hiperplasia folicular del centro germinal y distinguirla del timo normal.[78][79]
Otras pruebas que se deben considerar
La "prueba de la bolsa de hielo" puede ser una prueba diagnóstica útil para un paciente con una ptosis importante. El enfriamiento del párpado durante 2 a 5 minutos con una bolsa de hielo mejora la ptosis en >95% de los pacientes con MG, pero puede no mejorar la ptosis grave.[70][71]
La prueba del edrofonio (tensilón) es en gran medida de interés histórico, pero puede ser utilizada ocasionalmente por neuro-oftalmólogos y neurólogos para evaluar la oftalmoparesia y/o la ptosis aislada.
Los pacientes con ciertas formas de MG congénita no siempre se presentan en la infancia o en la niñez temprana. Suelen ser seronegativos (hay informes infrecuentes de anticuerpos MuSK en algunas familias), pero pueden tener pruebas neurofisiológicas anormales, a veces diferentes, pero en algunos tipos idénticas, a las de la MG autoinmunitaria. Si hay dudas, pueden realizarse pruebas genéticas para los síndromes miasténicos congénitos.
Crisis miasténica
La crisis miasténica es una emergencia médica. Se define como una exacerbación de la MG que requiere ventilación mecánica. Las indicaciones de ventilación mecánica son una capacidad vital forzada (CVF) de 15 mL/kg o menos (normal ≥60 mL/kg) y/o una fuerza inspiratoria negativa (FIN) de 20 cm H₂O o menos (normal ≥70 cm H₂O). Se toman mediciones en serie de la CVF y la FIN.
Es necesario un alto índice de sospecha clínica para las crisis miasténicas porque los pacientes pueden parecer inicialmente estables.[23][80] Los médicos no deben esperar el resultado anormal de una gasometría arterial (GSA), ya que se produce hacia el final de la evolución, después de la descompensación clínica. El juicio clínico siempre debe complementar y, si es necesario, sustituir los parámetros respiratorios.[81]
La crisis miasténica puede ser provocada por infecciones (en particular, infecciones respiratorias), aspiración, fármacos, incluyendo dosis altas de corticosteroides, cirugía, fármacos contraindicados o relativamente contraindicados en la MG, incumplimiento del tratamiento farmacológico, administración de inhibidores del punto de control inmunitario como terapia del cáncer o traumatismos.[57][58][80][81][82] Los pacientes de edad avanzada y los que también padecen otras enfermedades autoinmunitarias parecen presentar un mayor riesgo de desarrollar una crisis miasténica.[83]
Se deben tomar las medidas adecuadas para eliminar los factores desencadenantes. La debilidad muscular orofaríngea también puede contribuir a la insuficiencia respiratoria.
Cómo tomar una muestra de sangre venosa de la fosa antecubital utilizando una aguja de vacío.
El uso de este contenido está sujeto a nuestra cláusula de exención de responsabilidad