Etiologia
A deficiência de arginina-vasopressina (AVP-D; antes conhecida como diabetes insípido central) decorre de uma deficiência absoluta ou relativa de arginina-vasopressina (AVP); a resistência à AVP (AVP-R; antes conhecida como diabetes insípido nefrogênico) resulta de uma insensibilidade renal ou resistência à AVP. Ambos os mecanismos reduzem a permeabilidade dos dutos dentro do néfron à água, reduzindo a reabsorção de água e aumentando a perda de água pelos rins. As causas de AVP-D ou AVP-R podem ser genéticas ou adquiridas.[7]
AVP-D - adquirida
Cirurgia da hipófise: a cirurgia hipofisária transesfenoidal, geralmente para adenoma hipofisário, é uma causa comum de AVP-D.[3] Os adenomas hipofisários não apresentam AVP-D; a síndrome só se manifesta após intervenção cirúrgica. Caso a AVP-D ocorra na presença de uma massa selar detectada por exames de imagem, deve-se considerar um diagnóstico alternativo, como craniofaringioma, germinoma, metástases hipofisárias ou um processo granulomatoso.[3] A AVP-D após a cirurgia hipofisária pode ser transitória ou permanente. Raramente pode se manifestar como uma resposta trifásica: AVP-D inicial aguda, seguida de uma fase antidiurética transitória (apresentando-se como hiponatremia devido à síndrome de antidiurese inapropriada [SIAD]), evoluindo posteriormente para uma AVP-D permanente.[12]
Craniofaringioma: em contraste com a maioria dos tumores intracranianos, a AVP-D é relativamente comum em pacientes com craniofaringioma no pré (8 a 35%) e no pós-operatório (70 a 90%).[13] Pacientes com craniofaringiomas também têm maior probabilidade de apresentar anormalidades da sede primárias e pós-operatórias associadas, o que pode agravar o equilíbrio hídrico e os problemas eletrolíticos.[14]
Traumatismo cranioencefálico pós-traumático: cerca de 20% das lesões cerebrais traumáticas moderadas a graves são complicadas pela AVP-D.[3] Embora a maioria dos casos se resolva espontaneamente, a AVP-D permanente foi relatada em aproximadamente 7% dos casos.[15][16]
Lesões do pedúnculo hipofisário: o envolvimento do pedúnculo hipofisário é comum na histiocitose das células de Langerhans (HCL); em um estudo, a AVP-D foi relatada em até 24% dos pacientes com HCL de início pediátrico.[17] Outras condições infiltrativas que afetam o pedúnculo hipofisário e podem causar AVP-D incluem germinoma, metástases intracranianas (particularmente de tumores primários de mama ou pulmão), linfoma/leucemia, doenças granulomatosas (por exemplo, sarcoidose e tuberculose), infundíbulo-hipofisite linfocítica e doenças relacionadas à IgG4.[1][3][18][19][20][21][22][23][24][25] Crianças e adultos jovens com AVP-D idiopática e que apresentam espessamento do pedúnculo hipofisário no momento do diagnóstico, frequentemente desenvolvem um diagnóstico subjacente definido ao longo do tempo, conforme revelado pelo acompanhamento em longo prazo.[26][27]
Doenças autoimunes: a AVP-D está associada a outras doenças autoimunes endócrinas, incluindo tireoidite de Hashimoto, diabetes mellitus do tipo 1 e lúpus eritematoso sistêmico.[3][28] Os autoanticorpos contra as células secretoras de arginina-vasopressina (AVPcAb) foram relatados em 33% dos pacientes com AVP-D idiopática (não estrutural).[28]
Infecção do sistema nervoso central: a AVP-D pode se desenvolver como uma complicação tardia de meningite ou encefalite.[4][21][22][29][30]
A hemorragia subaracnoide envolvendo a artéria comunicante anterior (que fornece suprimento ao hipotálamo anterior) pode resultar em AVP-D e anormalidades da sede.[31] A AVP-D ocorre em 15% dos casos de hemorragia subaracnoide não traumática.[3]
Medicamentos: a fenitoína é uma possível causa de AVP-D, assim como a temozolomida (um medicamento quimioterápico oral usado primariamente para tratar certos tipos de tumor cerebral).[32][33]
A doença do coronavírus de 2019 (COVID-19) foi associada a raros casos de AVP-D inicial em um pequeno número de relatos de caso.[3][34][35] Os mecanismos propostos incluem a invasão viral direta do hipotálamo ou da hipófise posterior através dos receptores ACE2, a inflamação mediada imunologicamente que danifica os neurônios produtores de AVP e a lesão vascular ou hipóxica do eixo neuro-hipofisário durante doenças graves.[36][37] Em alguns casos, a condição parece transitória, sugerindo uma disfunção reversível.
A intoxicação por monóxido de carbono é uma causa rara de AVP-D.[38]
AVP-D - congênito/hereditário
AVP-D familiar representa aproximadamente 1% a 5% de todos os casos de AVP-D. Geralmente, é uma condição hereditária autossômica dominante devido a uma mutação no gene AVP-NPII localizado no cromossomo 20p135. Essa condição muitas vezes tem muitos familiares com AVP-D clínica precoce na vida.[39]
Malformações congênitas que envolvem a hipófise ou o hipotálamo e defeitos na linha média do prosencéfalo podem resultar em AVP-D.[40] Exemplos incluem displasia septo-óptica, agenesia do corpo caloso, síndrome da sela vazia e hipoplasia hipofisária.[3]
A AVP-D pode ocorrer como componente da síndrome de Wolfram (também chamada de síndrome DIDMOAD [diabetes insípido, diabetes mellitus, atrofia óptica e surdez]), um distúrbio neurodegenerativo autossômico recessivo e progressivo caracterizado por diabetes mellitus e atrofia óptica, com frequência variável de AVP-D e surdez neurossensorial.[41] Estudos genéticos identificaram dois subtipos de síndrome de Wolfram.[42] A WS1 é causada por mutações da perda de função no gene WFSI no cromossomo 4p16. O WFSI codifica uma glicoproteína de 890 aminoácidos, wolframina, que regula o estresse do retículo endoplasmático e a homeostase do cálcio. A perda de função da wolframina desencadeia apoptose e morte celular. Mutações não inativadoras do WFSI estão associadas à perda auditiva neurossensorial autossômica dominante. A WSI pode, portanto, representar um extremo de um espectro de um distúrbio. A WS2 também é caracterizada por atrofia óptica e diabetes mellitus. A WS2 é causada por mutações de perda de função no gene CISD2, que codifica uma proteína expressa no retículo endoplasmático e na membrana mitocondrial externa. A perda de função do CISD2 interrompe o fluxo de cálcio, levando à autofagia e à morte celular.[41]
AVP-R - adquirida
Induzido por medicamentos: o lítio é uma causa bem estabelecida de AVP-R, com estudos relatando sua ocorrência em até 40% dos pacientes que recebem terapia prolongada com lítio, e algumas estimativas sugerindo taxas de até 85%.[9][10] O mecanismo envolve a disrupção, pelo lítio, dos canais de aquaporina-2 (AQP2) nos ductos coletores renais. A AVP-R pode ser irreversível, persistindo mesmo após a interrupção da terapia com lítio.[43] Outros medicamentos que podem desencadear AVP-R incluem demeclociclina, cisplatina, colchicina, gentamicina, rifampicina e sevoflurano.[5][44]
Doença sistêmica, desequilíbrio eletrolítico e uropatia pós-obstrutiva: doença renal crônica, sarcoidose renal e amiloidose renal são causas reconhecidas de AVP-R.[5][20] A AVP-R também pode ser causada por hipercalcemia e hipocalemia; nesses casos, pode ser reversível com a correção do desequilíbrio eletrolítico subjacente.[5] A obstrução ureteral é outro fator de risco reconhecido para AVP-R.[5] Acredita-se que o aumento da pressão hidrostática suprima a expressão do canal AQP2 nos ductos coletores renais. Essa down-regulation pode persistir por até 30 dias após o alívio da obstrução, o que pode explicar a diurese frequentemente observada após a liberação de obstrução grave do trato urinário.[10]
AVP-R - hereditária
Aproximadamente 90% dos casos familiares de AVP-R são devidos a mutações de perda de função no gene AVPR2, que codifica o receptor V2 da vasopressina, responsável por mediar os efeitos antidiuréticos da AVP nos ductos coletores renais. Essas mutações resultam em herança recessiva ligada ao cromossomo X.[5][10][45]
A AQP2 é o canal de água dependente de AVP que torna o duto coletor distal permeável à água e permite a reabsorção de água renal distal mediada por AVP. Mutações de perda de função no gene AQP2 produzem AVP-R familiar autossômico recessivo.[5][10][45] Mutações de perda de função no gene transportador de ureia-B também produzem AVP-R autossômica recessiva.[39]
Gestação e AVP-D
Os limiares osmóticos para sede e liberação de AVP são alterados na gravidez.[46] Também existe um aumento de quatro vezes no clearance metabólico de AVP devido à produção placentária de vasopressinase/oxitocinase. Isso pode desmascarar AVP-D não reconhecida anteriormente. Além disso, a gestação pode agravar a gravidade da AVP-R ou da AVP-D preexistente.[1][6][47] A AVP-D transitória gestacional foi relatada em pacientes com pré-eclâmpsia, síndrome HELLP (hemólise, enzimas hepáticas elevadas e plaquetopenia) e doença hepática gordurosa associada a disfunção metabólica aguda (anteriormente conhecida como doença hepática gordurosa não alcoólica), onde a degradação hepática prejudicada da vasopressinase é um fator contribuinte.
Fisiopatologia
O volume plasmático e a osmolaridade são mantidos em uma faixa estreita por um sistema neuro-humoral integrado que coordena o equilíbrio entre o consumo dirigido pela sede (ingestão de líquidos) e a perda renal de água (perda de líquidos).[48][49]
O principal regulador da perda de água renal é o hormônio peptídeo de 9 aminoácidos arginina-vasopressina (AVP), também conhecido como hormônio antidiurético (HAD).[48] O AVP é sintetizado em neurônios magnocelulares nos núcleos supraótico (SON) e paraventricular (PVN) do hipotálamo. O AVP é produzido a partir de um grande peptídeo precursor. A transcrição primária passa por um processamento pós-tradução significativo, pois é trafegada pelos axônios dos neurônios magnocelulares que terminam na hipófise posterior, onde o hormônio é armazenado e liberado. Cada molécula de AVP é co-secretada com dois fragmentos do precursor original: copeptina e neurofisina II (NPII).[50] O processamento é descrito esquematicamente abaixo.
[Figure caption and citation for the preceding image starts]: Vasopressina: estrutura gênica e processamento pós-traduçãoDo acervo pessoal do Dr Stephen Ball [Citation ends].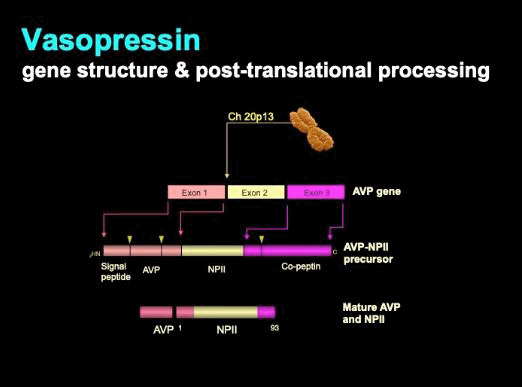
Existe uma relação linear entre a concentração de AVP no plasma e a osmolaridade no plasma. A osmolalidade plasmática é detectada pelos osmorreceptores no hipotálamo, e esses aferentes sensoriais regulam a síntese e liberação de AVP. A liberação de AVP também é regulada pelo volume circulante e pressão arterial. Essa barorregulação pode levar à produção e liberação de AVP nas osmolalidades plasmáticas abaixo do limiar osmolar padrão de cerca de 290 mmol/kg (290 mOsm/kg).[48][49]
[Figure caption and citation for the preceding image starts]: Fisiologia da AVP e da sedeDo acervo pessoal do Dr Stephen Ball [Citation ends].
A estimulação hiperosmolar graduada define muito claramente as características fisiológicas da produção de AVP e sede osmorreguladas, representadas nessas duas figuras. A relação linear entre osmolaridade plasmática e AVP plasmática é ainda caracterizada por um limiar osmolar funcional para liberação de AVP; e uma inclinação característica, que serve para definir a sensibilidade da resposta do AVP à estimulação osmolar. Um conjunto muito semelhante de características descreve a relação da sede com a osmolaridade plasmática. Embora as características de liberação de AVP e da sede osmorreguladas variem entre os indivíduos (o intervalo descrito aqui como áreas sombreadas ou nomograma), as características permanecem notavelmente constantes dentro de um indivíduo (representadas aqui como a linha sólida dentro do nomograma) ao longo do tempo.
A AVP atua nos receptores de AVP do tipo 2 (AVPR2) na superfície intersticial das células do ducto coletor distal renal, aumentando a permeabilidade à água através da síntese aumentada dos canais de água aquaporina-2.[48] Estes são montados e inseridos na membrana apical das células do ducto coletor, resultando na reabsorção da água dependente de AVP e na concentração de urina.
AVP-D resulta de qualquer afecção que prejudique a produção, o transporte ou a liberação de AVP.
Estima-se que a destruição de mais de 90% dos neurônios vasopressinérgicos no hipotálamo/hipófise posterior seja necessária para esgotar a AVP o suficiente para causar poliúria hipotônica.[3]
AVP-R resulta de afecções que prejudicam a capacidade dos dutos coletores renais de responder à AVP.
Tanto a AVP-D quanto a AVP-R são caracterizadas por comprometimento da reabsorção renal de água, resultando na produção excessiva de urina hipotônica (diluída) (poliúria), tipicamente superior a 3 litros (ou >50 mL/kg) por dia.[3] Isso é acompanhado por uma sede e aumento do consumo de líquidos (polidipsia) significativos, pois os osmorreceptores centrais e barorreceptores periféricos estimulam a sede central e os comportamentos dependentes da sede para manter o volume circulante e o status osmolar.
Os sintomas de pacientes com AVP-D ou AVP-R decorrente de uma etiologia não traumática geralmente têm um início insidioso. Pacientes com AVP-D após lesão cerebral traumática ou cirurgia da hipófise geralmente apresentam um início mais rápido dos sintomas. A gravidade da poliúria na AVP-D corresponde à extensão do dano neuronal, sendo que a destruição completa causa deficiência total de AVP e poliúria acentuada, enquanto o dano parcial com secreção residual de AVP resulta em AVP-D parcial e sintomas mais leves.[3]
A gestação está associada a diversas mudanças na regulação de sal e água.[46] AVP-D transitória pode se desenvolver como uma consequência da diminuição do limiar osmótico para a sede e para a liberação de AVP, além de uma diminuição na osmolalidade plasmática. Há também um aumento de quatro vezes no clearance metabólico da AVP devido à produção placentária de vasopressinase/oxitocinase no segundo ou terceiro trimestre.[3] Além disso, a gestação pode agravar a gravidade de AVP-D ou AVP-R preexistente.[1][6][47]
Classificação
Síndromes de poliúria-polidipsia
Polidipsia primária:
Um distúrbio caracterizado por uma sensação anormalmente intensa de sede, não causada por fatores fisiológicos como desidratação ou disfunção da AVP. É principalmente uma condição de aumento da sensação de sede, frequentemente ligada a fatores psicológicos ou psiquiátricos e, ocasionalmente, associada a anormalidades estruturais no cérebro. Os pacientes geralmente relatam ingestão excessiva de líquidos, seguida de poliúria. Essa condição geralmente indica uma perturbação na regulação da sede, e não um defeito na secreção ou função da AVP.
Pode ser produzido por medicamentos que causam xerostomia, estar associado a síndromes psiquiátricas ou ser habitual.[1]
A polidipsia psicogênica é um subtipo de polidipsia primária, caracterizada pela ingestão excessiva de líquidos desencadeada por condições psiquiátricas. É mais comum em pacientes com esquizofrenia, transtornos de ansiedade ou outros transtornos mentais.
AVP-D:
Decorrente da síntese ou liberação deficiente de arginina-vasopressina (AVP) no eixo hipotálamo-hipófise.[1]
Pode ser congênito (hereditário) ou adquirido (por exemplo, procedimentos neurocirúrgicos para tumores, trauma, autoimune, infiltrante, vascular).[3][4]
AVP-R:
Decorrente de insensibilidade renal ou resistência à AVP, com a resultante falta de permeabilidade do duto coletor à água.[1]
Pode ser congênito (hereditário) ou adquirido (por exemplo, induzida por medicamentos, particularmente lítio; hipercalcemia, hipocalemia; uropatia obstrutiva).[5]
A AVP-D ou a AVP-R transitória na gestação (antes conhecida como diabetes gestacional insípido) pode ocorrer por causa do metabolismo acelerado de AVP causado pela produção placentária de aminopeptidases de cisteína.[6]
O uso deste conteúdo está sujeito ao nosso aviso legal