Etiologia
A doença de Lesch-Nyhan (LND) é causada por uma mutação no gene que codifica a enzima hipoxantina-guanina fosforibosiltransferase (HPRT) no braço longo do cromossomo X, no Xq26-q27.[11] As mutações são heterogêneas, incluindo mutações pontuais e outras substituições, deleções e inserções.[12][13] Acredita-se geralmente que a maioria das mutações ocorre de novo, porque homens com LND geralmente não têm filhos devido aos efeitos debilitantes da doença. As mutações que causam a doença aparecem em todo o gene HPRT, com alguns "hotspots" mutacionais menores. As correlações genótipo-fenótipo não indicam que características específicas da doença estejam associadas a sítios específicos de mutação. Em geral, contudo, manifestações clínicas menos graves (variantes de Lesch-Nyhan [LNV]) resultam de mutações preditas a permitirem algum grau de função enzimática residual.[12][13] GeneReviews: Lesch-Nyhan syndrome Opens in new window
Fisiopatologia
A HPRT media a reciclagem de hipoxantina e guanina em seus respectivos pools de nucleotídeos. Na ausência de HPRT, a hipoxantina e a guanina não são recicladas, mas degradadas em ácido úrico. O salvamento reduzido de purinas, juntamente com a ativação de novo da síntese de purinas, causa uma acentuada superprodução de ácido úrico.[3] Apesar dessa superprodução, um clearance renal eficiente limita os níveis médios de ácido úrico sérico, cujo aumento é tipicamente menor que duas vezes. A excreção total de ácido úrico renal em pacientes com a LND clássica é tipicamente quatro vezes maior que a dos controles. Valores crônicos de urato sérico >416 micromoles/L (>7.0 mg/dL) são associados com aumento do risco de deposição de cristais de urato nas articulações, nos rins e nos tecidos subcutâneos.[14] Se não tratada, pode causar artrite gotosa, nefrolitíase e tofos subcutâneos.
A LND é acompanhada por uma intensa diminuição no conteúdo de dopamina nos gânglios da base do cérebro, o que se acredita ser um determinante importante das características neurocomportamentais, incluindo o distúrbio motor hipercinético, os deficits de atenção e o comportamento anormal.[4][15][16] Além disso, a LND e LNV são associadas com anomalias extensivas de substância cinzenta e substância branca, que também podem oferecer pistas importantes sobre substratos neurais do fenótipo.[17][18][19] A relação exata entre deficiência de HPRT e disfunção da dopamina na LND ainda não está clara. Entretanto, evidências sugerem uma relação importante entre vias recicladoras de purina e a integridade neuroquímica do fenótipo dopaminérgico, tendo sido sugerida uma função para HPRT nos processos de neurodesenvolvimento.[20][21][22][23] Apesar da deficiência de dopamina, o efeito da terapia de reposição de dopamina em pacientes com fenótipos estabelecidos é inconsistente e geralmente inútil, presumivelmente devido ao desenvolvimento de supersensibilidade do receptor de dopamina e outras alterações de neuroplasticidade.[3][24] Um estudo de 2024 relatou que iniciar a terapia de reposição de dopamina muito cedo na vida (ou seja, antes do desenvolvimento completo do fenótipo) pode prevenir parcialmente essas mudanças plásticas para limitar os sintomas, embora mais pesquisas sejam necessárias.[25] Alguns pacientes também desenvolvem espasticidade e hiper-reflexia, indicativas de disfunção no sistema motor corticoespinhal, que podem resultar de mielopatia causada por movimentos involuntários do pescoço crônicos e fortes.[26]
GeneReviews: Lesch-Nyhan syndrome
Opens in new window[Figure caption and citation for the preceding image starts]: Papel da enzima hipoxantina-guanina fosforibosiltransferase (HPRT) no grande esquema do metabolismo da purinaCriado por J.E. Visser, MD, PhD e H.A. Jinnah, MD, PhD; usado com permissão [Citation ends].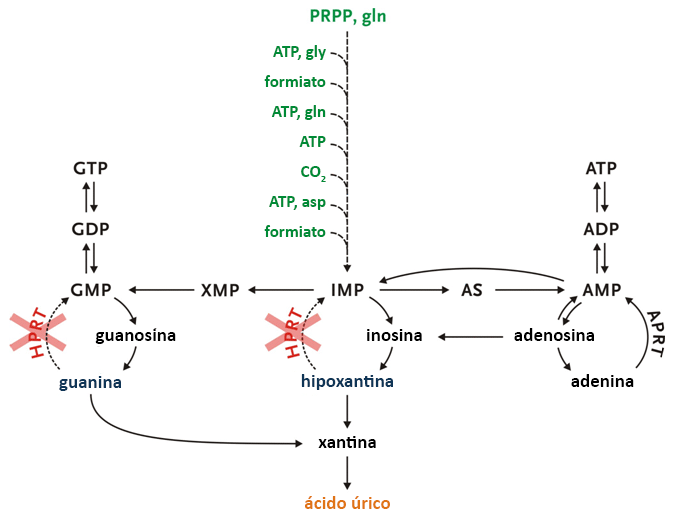 [Figure caption and citation for the preceding image starts]: Níveis de ácido úrico em pacientes com a doença de Lesch-Nyhan (LND) clássica, pacientes com variantes de Lesch-Nyhan (LNV) e controles saudáveis. SD, desvio-padrão; HRH: hiperuricemia relacionada à hipoxantina-guanina fosforibosiltransferase (HPRT); HRD: doença neurológica relacionada à HPRTDo acervo de J.E. Visser, MD, PhD e H.A. Jinnah, MD, PhD; usado com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Níveis de ácido úrico em pacientes com a doença de Lesch-Nyhan (LND) clássica, pacientes com variantes de Lesch-Nyhan (LNV) e controles saudáveis. SD, desvio-padrão; HRH: hiperuricemia relacionada à hipoxantina-guanina fosforibosiltransferase (HPRT); HRD: doença neurológica relacionada à HPRTDo acervo de J.E. Visser, MD, PhD e H.A. Jinnah, MD, PhD; usado com permissão [Citation ends].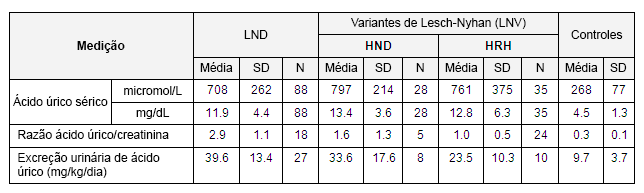
Classificação
LND, LNV: doença neurológica e hiperuricemia relacionadas à HPRT[3][5][6]
A HPRT é associada a um continuum fenotípico, no qual a ocorrência e a intensidade das características clínicas são dependentes da atividade enzimática residual.
Pacientes com LND clássica, virtualmente sem atividade enzimática, têm o fenótipo completo: hiperuricemia, disfunção neurológica, deficits cognitivos e comportamento anormal, incluindo autolesão.
LNV, com alguma atividade residual da HPRT, têm um fenótipo parcial: não apresentam comportamento autolesivo, podem apresentar hiperuricemia com graus variados de disfunção motora e cognitiva ou ter apenas hiperuricemia.[5] As LNVs são tipicamente subdivididas em:
Doença neurológica relacionada à HPRT (HND): hiperuricemia e algum grau de disfunção neurológica e/ou deficits cognitivos.
Hiperuricemia relacionada à HPRT (HRH): somente hiperuricemia, sem disfunção neurológica.
O epônimo síndrome de Kelley-Seegmiller também foi usado para as LNVs.[7] No entanto, é melhor evitar esse termo; sua definição não é clara, pois ele tem sido usado para se referir a todas as variantes ou apenas àquelas com hiperuricemia isolada. Além disso, ele sugere outra entidade nosológica, mas é meramente uma variante da LND.[Figure caption and citation for the preceding image starts]: Espectro fenotípico associado com a deficiência de hipoxantina-guanina fosforibosiltransferase (HPRT). HRH: Hiperuricemia relacionada à HPRT; HND: Doença neurológica relacionada à HPRT; LND: doença de Lesch-NyhanDo acervo de J.E. Visser, MD, PhD e H.A. Jinnah, MD, PhD; usado com permissão [Citation ends].
O uso deste conteúdo está sujeito ao nosso aviso legal