Etiologia
Deficiência de hormônio do crescimento (DGH) congênita
Inclui um grupo em expansão de diferentes distúrbios etiológicos.[5][6] Ela pode ser isolada ou ocorrer com deficiências hormonais hipofisárias combinadas (DHHC), ou com características extra-hipofisárias, como a hipoplasia do nervo óptico, defeitos na linha média do prosencéfalo, deficiência intelectual, anormalidades no pescoço, anormalidades no cerebelo ou holoprosencefalia (hipopituitarismo sindrômico). A maioria dos casos é de origem idiopática.
Complicações perinatais, como prematuridade, sangramento gestacional, parto complicado e sofrimento ou asfixia fetal também podem ser fatores de risco.[7] No entanto, há controvérsias se esses são verdadeiros fatores de risco ou complicações do hipopituitarismo.
A DGH congênita pode ser esporádica ou familiar em 5% a 30% dos casos. Uma lista em rápida expansão de mutações em vários fatores de transcrição e genes da hipófise foi identificada para DGH e DHHC (por exemplo, GHI, GHRHR, PROP1, POU1F1, HESX1, LHX3, LHX4, SOX3, OTX2 e RNPC3). A penetrância variável e a herança oligogênica são comuns e, portanto, a interpretação dos achados genéticos requer a participação de especialistas.[1][8][9][10][11][12]
Há 6 tipos definidos de DGH familiar isolada (DIGH).[1]
[Figure caption and citation for the preceding image starts]: Tipos de síndromes familiares de deficiência isolada de hormônio do crescimento. DHHC, deficiências hormonais hipofisárias combinadas; GH, hormônio do crescimento; DIGH, deficiência isolada de hormônio do crescimento; HCHr, hormônio do crescimento humano recombinante; TSH, hormônio estimulante da tireoide; PRL, prolactinaCriado pelo BMJ Knowledge Centre com base nas referências citadas no tópico [Citation ends].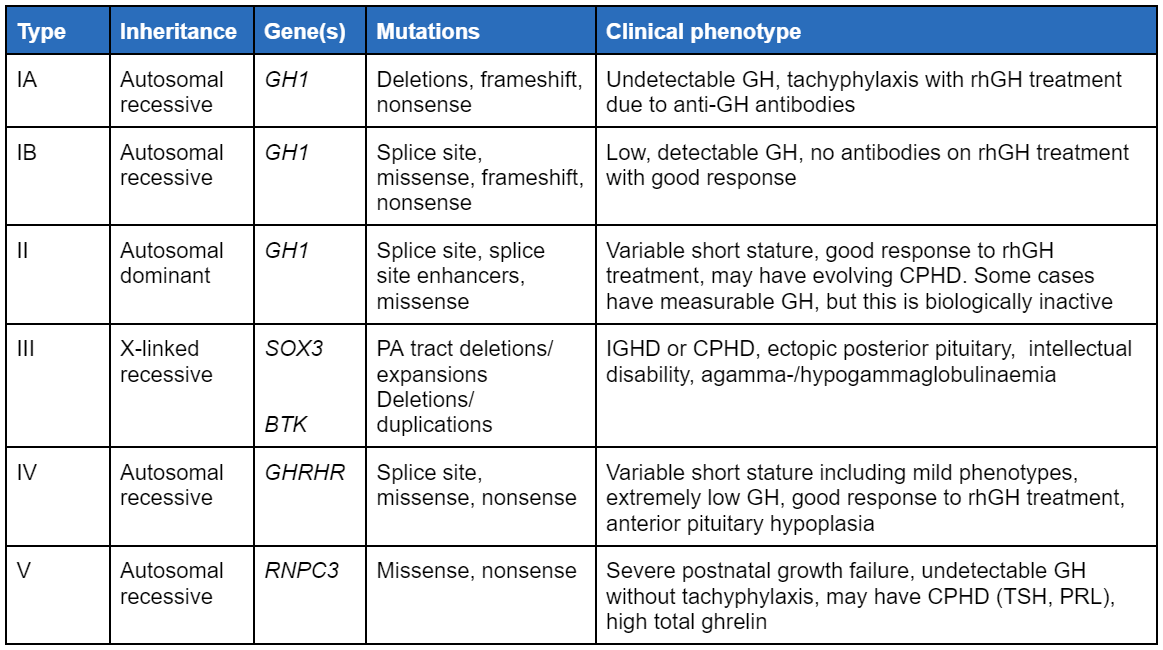
DGH adquirida
Tumores hipotálamo-hipofisários: podem ser benignos ou malignos, císticos ou sólidos. A DGH pode surgir por causa de um tumor, como resultado de uma cirurgia ou após radioterapia. O tumor supra/intrasselar mais comum na infância é o craniofaringioma.[13][14][15] Outros tumores raros observados nessa região incluem adenomas hipofisários, tumores de células germinativas, gliomas da via óptica, hamartomas, meningiomas e cordomas. Lesões císticas, como cistos da fenda de Rathke, cistos aracnoides ou cistos dermoides e, ainda mais raramente, tumores metastáticos, como carcinomas nasofaríngeos e de Hodgkin na infância, também podem causar a DGH.
Radioterapia para tumores do sistema nervoso central (SNC): a exposição da região hipotálamo-hipofisária a qualquer irradiação (local e distalmente; por exemplo, meduloblastomas, ependimoma ou para neoplasias hematológicas e transplante de medula óssea) pode resultar em DGH profunda.[16]
Lesão cerebral traumática: dano hipofisário pode ocorrer após lesão cerebral traumática (por exemplo, acidentes de trânsito, lesão não acidental) ou após neurocirurgia.[17][18]
Outras causas raras incluem infecções (por exemplo, meningite, encefalite, abscesso hipofisário); distúrbios inflamatórios/infiltrativos (por exemplo, sarcoidose, tuberculose, hipofisite linfocítica autoimune, histiocitose das células de Langerhans); distúrbios de sobrecarga de ferro (por exemplo, hemocromatose primária, talassemia e outras doenças que requerem transfusões crônicas).[19]
Uma DGH reversível também pode surgir como resultado de uma privação psicossocial.[20]
Fisiopatologia
A hipófise inclui a adeno-hipófise (lobos anterior e intermediário) e a neuro-hipófise (lobo posterior). A adeno-hipófise consiste em somatotrofos que produzem o GH, tireotrofos que secretam a tireotrofina ou o hormônio estimulante da tireoide (TSH), corticotrofos que secretam a corticotrofina ou o hormônio adrenocorticotrófico (ACTH), gonadotrofos que produzem o hormônio folículo-estimulante (FSH) e o hormônio luteinizante (LH), e lactotrofos que secretam a prolactina.
O hormônio de liberação do hormônio do crescimento (GHRH) hipotalâmico estimula a liberação de GH, e a somatostatina inibe essa liberação. A liberação de GH é pulsátil e ocorre particularmente durante o sono de ondas lentas, com pulsos de maior amplitude liberados na infância do que na idade adulta. A função da grelina endógena no crescimento normal não é conhecida.[21][22] O gene GH humano (GH) faz parte de um conjunto de 5 genes homólogos e está localizado no braço longo do cromossomo 17 (17q22-24).
O GH secretado pelos somatotrofos da hipófise liga-se aos seus receptores e ativa uma cascata de sinalização que conduz finalmente à geração do fator de crescimento semelhante à insulina-1 (IGF-1) que, em seguida, medeia as várias ações de promoção de crescimento do GH. As concentrações de IGF-1 apresentam uma boa correlação com aquelas do GH. No entanto, baixos níveis de IGF-1 podem ser observados no hipotireoidismo, na desnutrição, no diabetes mellitus mal controlado e nas doenças crônicas. Crianças com DGH não tratadas serão baixas e poderão ter puberdade tardia, diminuição do estirão de crescimento puberal e um escore de desvio-padrão (EDP) da estatura final de -4 a -6.[21][22][23] GH também aumenta a absorção de cálcio e a densidade óssea, é lipolítico, reduz a gordura corporal e aumenta a massa muscular.
Alguns dados sugerem que as crianças com DGH isolada possam ter comprometimento cognitivo, incluindo escores mais baixos de QI, Índice de Compreensão Verbal, Índice de Velocidade de Processamento e Movement Assessment Battery for Children (bateria para avaliação do movimento de crianças).[24] No entanto, estudos adicionais são necessários para investigar os efeitos e impactos da DGH na estrutura do cérebro, na função cognitiva e no desempenho motor.
Os somatotrofos constituem 40% a 50% da população de células da hipófise e são especialmente vulneráveis aos efeitos da pressão e à lesão por radiação. A rica rede vascular do hipotálamo e da hipófise e a estrutura do pedúnculo hipofisário torna-o vulnerável aos efeitos de um trauma. Como resultado, a DGH e o deficit de crescimento são manifestações endócrinas precoces, seguidas de deficiência de gonadotrofina e TSH, após qualquer injúria à região do hipotálamo-hipófise. Tumores e outras lesões na área do hipotálamo-hipófise podem causar distúrbios endócrinos diretamente ou secundários ao tratamento (cirurgia, radioterapia).
Classificação
Classificação etiológica
Não existe uma classificação formal, além da classificação para DGH isolada (DIGH), que é baseada no genótipo.[1] A deficiência de hormônio do crescimento (DGH) pode estar presente desde o nascimento (embora possa ser evidente apenas na primeira infância ou adolescência) ou pode ser adquirida. Ela também pode ser classificada como DIGH ou como deficiência hormonal hipofisária combinada (DHHC). A DIGH congênita pode ser esporádica ou familiar; 5% a 30% são familiares e existem 6 tipos definidos (ver tabela na seção Etiologia).
O uso deste conteúdo está sujeito ao nosso aviso legal