Nos casos suspeitos, deve-se realizar uma avaliação com história (incluindo história familiar), exame físico, ECG e ecocardiografia. Esta última estabelece o diagnóstico. Os pacientes assintomáticos geralmente são diagnosticados no momento de um exame cardíaco de rotina ou de um rastreamento familiar.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
[4]Wigle ED, Rakowski H, Kimball BP, et al. Hypertrophic cardiomyopathy: clinical spectrum and treatment. Circulation. 1995 Oct 1;92(7):1680-92.
https://www.ahajournals.org/doi/10.1161/01.CIR.92.7.1680
http://www.ncbi.nlm.nih.gov/pubmed/7671349?tool=bestpractice.com
Os pacientes são mais frequentemente diagnosticados após o início das manifestações clínicas, no entanto, com apenas 32% dos pacientes sendo diagnosticados na avaliação médica de rotina.[21]Adabag AS, Kuskowski MA, Maron BJ. Determinants for clinical diagnosis of hypertrophic cardiomyopathy. Am J Cardiol. 2006 Dec 1;98(11):1507-11.
http://www.ncbi.nlm.nih.gov/pubmed/17126660?tool=bestpractice.com
A análise laboratorial do DNA para genes mutantes é o método mais definitivo para estabelecer o diagnóstico de cardiomiopatia hipertrófica (CMH), mas o método costuma ser usado apenas para fins de rastreamento.
História
História familiar
Uma história familiar de síncope, insuficiência cardíaca ou morte prematura ou súbita deve ser considerada. Às vezes, as mortes súbitas cardíacas podem ter sido relatadas como mortes acidentais, por exemplo, afogamento ou acidentes de trânsito inexplicados.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Outros pontos a serem observados na história familiar incluem transplante cardíaco, implantes de marca-passo e desfibrilador, e características sugestivas de doença sistêmica (por exemplo, AVC em pouca idade, fraqueza muscular esquelética ou doença renal). Uma árvore genealógica de três a quatro gerações deve ser criada para auxiliar no diagnóstico, fornecer pistas sobre a etiologia, determinar o padrão de herança e identificar parentes em risco.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
A CMH sarcomérica é autossômica dominante e, portanto, caracterizada pela presença de indivíduos afetados ao longo das gerações, com transmissão de pais de ambos os sexos e um risco de 50% de transmissão de alelos para os descendentes. A história familiar pode ser negativa, no entanto, pois a doença tem uma penetrância incompleta.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Sintomas
Os pacientes podem ser assintomáticos. Entretanto, é importante notar qualquer sintoma de pré-síncope ou síncope, principalmente quando ocorre com exercícios, dispneia aos esforços, palpitações ou dor torácica. Pacientes acima dos 50 anos de idade podem apresentar fibrilação atrial ou sintomas de acidente vascular cerebral (AVC).[22]Robinson K, Frenneaux MP, Stockins B, et al. Atrial fibrillation in hypertrophic cardiomyopathy: a longitudinal study. J Am Coll Cardiol. 1990 May;15(6):1279-85.
https://www.jacc.org/doi/pdf/10.1016/S0735-1097%2810%2980014-2
http://www.ncbi.nlm.nih.gov/pubmed/2329232?tool=bestpractice.com
Exame físico
O exame físico pode evidenciar uma elevação do ventrículo esquerdo (VE); um duplo impulso apical; um "upstroke" carotídeo rápido; um sopro sistólico de ejeção na borda esquerda inferior, que é acentuado por exercício e pela posição ortostática e atenuado na posição supina ou agachada; e uma quarta bulha cardíaca.[4]Wigle ED, Rakowski H, Kimball BP, et al. Hypertrophic cardiomyopathy: clinical spectrum and treatment. Circulation. 1995 Oct 1;92(7):1680-92.
https://www.ahajournals.org/doi/10.1161/01.CIR.92.7.1680
http://www.ncbi.nlm.nih.gov/pubmed/7671349?tool=bestpractice.com
Teste diagnóstico
eletrocardiograma (ECG)
Um ECG de 12 derivações em repouso é recomendado na primeira consulta clínica em todos os indivíduos com CMH conhecida ou suspeita e deve ser repetido sempre que houver alteração nos sintomas em pacientes com diagnóstico estabelecido.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
A maioria dos pacientes apresenta anormalidades eletrocardiográficas; elas não são específicas da CMH, mas devem levar a investigações adicionais com ecocardiografia. Um ECG anormal pode anteceder o achado de hipertrofia na ecocardiografia.[23]Yetman AT, McCrindle BW. Management of pediatric hypertrophic cardiomyopathy. Curr Opin Cardiol. 2005 Mar;20(2):80-3.
http://www.ncbi.nlm.nih.gov/pubmed/15711191?tool=bestpractice.com
Anormalidades de repolarização são comuns. A inversão da onda T comumente envolve as derivações inferior e lateral e as ondas T são profundas e frequentemente precedidas por infradesnivelamento do segmento ST.[24]Finocchiaro G, Sheikh N, Biagini E, et al. The electrocardiogram in the diagnosis and management of patients with hypertrophic cardiomyopathy. Heart Rhythm. 2020 Jan;17(1):142-51.
http://www.ncbi.nlm.nih.gov/pubmed/31349064?tool=bestpractice.com
Ondas T profundamente invertidas nas derivações precordiais são sugestivas de CMH apical.[25]Veselka J, Anavekar NS, Charron P. Hypertrophic obstructive cardiomyopathy. Lancet. 2017 Mar 25;389(10075):1253-67.
http://www.ncbi.nlm.nih.gov/pubmed/27912983?tool=bestpractice.com
Ondas Q anormais proeminentes podem ser observadas nas derivações inferiores (II, III, aVF) e/ou laterais (I, aVL, V5-6), refletindo hipertrofia septal.[25]Veselka J, Anavekar NS, Charron P. Hypertrophic obstructive cardiomyopathy. Lancet. 2017 Mar 25;389(10075):1253-67.
http://www.ncbi.nlm.nih.gov/pubmed/27912983?tool=bestpractice.com
Podem estar presentes voltagens elevadas do intervalo QRS indicando hipertrofia ventricular esquerda (HVE). Elas estão quase sempre associadas a outras anormalidades eletrocardiográficas na CMH.[24]Finocchiaro G, Sheikh N, Biagini E, et al. The electrocardiogram in the diagnosis and management of patients with hypertrophic cardiomyopathy. Heart Rhythm. 2020 Jan;17(1):142-51.
http://www.ncbi.nlm.nih.gov/pubmed/31349064?tool=bestpractice.com
A presença de critérios isolados de voltagem do intervalo QRS para HVE na ausência de outros marcadores de ECG está presente em menos de 2% dos pacientes com CMH.[26]Bernardini A, Crotti L, Olivotto I, et al. Diagnostic and prognostic electrocardiographic features in patients with hypertrophic cardiomyopathy. Eur Heart J Suppl. 2023 May;25(suppl c):C173-8.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10132576
http://www.ncbi.nlm.nih.gov/pubmed/37125268?tool=bestpractice.com
Podem ser observados sinais de ECG de aumento atrial esquerdo e direito e prolongamento da onda P (um conhecido preditor de fibrilação atrial). Raramente ocorrem isoladamente; outras anormalidades eletrocardiográficas, tais como alterações de repolarização ou sinais de HVE, estão geralmente presentes. O aumento do átrio esquerdo reflete disfunção diastólica, altas pressões de enchimento, obstrução do fluxo de saída e regurgitação mitral funcional. Dilatação e disfunção atrial esquerda são marcadores de prognóstico adverso.[24]Finocchiaro G, Sheikh N, Biagini E, et al. The electrocardiogram in the diagnosis and management of patients with hypertrophic cardiomyopathy. Heart Rhythm. 2020 Jan;17(1):142-51.
http://www.ncbi.nlm.nih.gov/pubmed/31349064?tool=bestpractice.com
Desvio do eixo esquerdo (causado por HVE) e pré-excitação ventricular também podem ser observados.[24]Finocchiaro G, Sheikh N, Biagini E, et al. The electrocardiogram in the diagnosis and management of patients with hypertrophic cardiomyopathy. Heart Rhythm. 2020 Jan;17(1):142-51.
http://www.ncbi.nlm.nih.gov/pubmed/31349064?tool=bestpractice.com
Alguns pacientes podem apresentar arritmias, por exemplo, fibrilação atrial ou taquicardia supraventricular.[24]Finocchiaro G, Sheikh N, Biagini E, et al. The electrocardiogram in the diagnosis and management of patients with hypertrophic cardiomyopathy. Heart Rhythm. 2020 Jan;17(1):142-51.
http://www.ncbi.nlm.nih.gov/pubmed/31349064?tool=bestpractice.com
O ECG é normal apenas em uma pequena proporção (5% a 10%) dos pacientes na apresentação.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
[25]Veselka J, Anavekar NS, Charron P. Hypertrophic obstructive cardiomyopathy. Lancet. 2017 Mar 25;389(10075):1253-67.
http://www.ncbi.nlm.nih.gov/pubmed/27912983?tool=bestpractice.com
Foi relatado que esses pacientes apresentam uma evolução clínica mais favorável do que aqueles com anormalidades eletrocardiográficas.[26]Bernardini A, Crotti L, Olivotto I, et al. Diagnostic and prognostic electrocardiographic features in patients with hypertrophic cardiomyopathy. Eur Heart J Suppl. 2023 May;25(suppl c):C173-8.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC10132576
http://www.ncbi.nlm.nih.gov/pubmed/37125268?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: Eletrocardiografia (ECG) mostrando alterações associadas à hipertrofia ventricular esquerda (HVE)Do acervo de Melanie Everitt, MD, Heart Failure & Transplantation Program, Primary Children's Medical Center, Salt Lake City, UT; usado com permissão [Citation ends].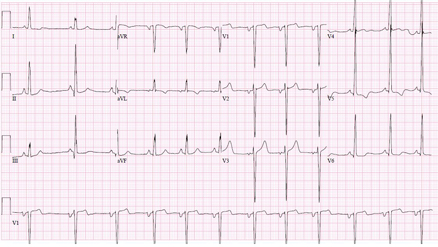 [Figure caption and citation for the preceding image starts]: Inversão de onda T giganteDo acervo do Dr Anji T. Yetman, MD, University of Utah [Citation ends].
[Figure caption and citation for the preceding image starts]: Inversão de onda T giganteDo acervo do Dr Anji T. Yetman, MD, University of Utah [Citation ends].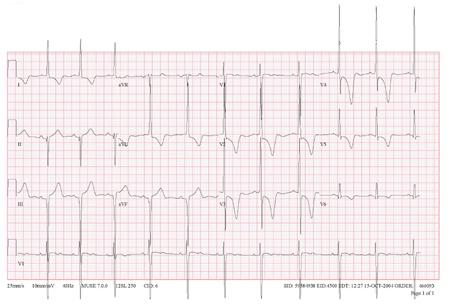
Exames laboratoriais
As diretrizes dos EUA não recomendam exames laboratoriais como parte da investigação inicial.[2]Writing Committee Members, Ommen SR, Ho CY, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy: a report of the American Heart Association/American College of Cardiology Joint Committee on clinical practice guidelines. J Am Coll Cardiol. 2024 Jun 11;83(23):2324-405.
https://www.sciencedirect.com/science/article/pii/S0735109724003826?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/38727647?tool=bestpractice.com
No entanto, as diretrizes europeias recomendam que todos os pacientes com suspeita ou confirmação de CMH sejam submetidos a exames laboratoriais de rotina para estabelecer a etiologia, avaliar a gravidade da doença, auxiliar na detecção de manifestações extracardíacas e na avaliação de disfunções orgânicas secundárias. Os seguintes testes de primeira linha são recomendados:[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Creatina quinase (CK): níveis elevados são uma pista útil ao tentar estabelecer a etiologia; distúrbios metabólicos como doença de Danon ou mitocondrial, que podem mimetizar a CMH, devem ser considerados. Quando a CK está persistentemente elevada, um exame detalhado por um neurologista deve ser considerado.
Testes da função hepática: a disfunção hepática é prevalente em pacientes com insuficiência cardíaca crônica. Testes da função hepática anormais também podem ser uma pista útil ao tentar estabelecer a etiologia; distúrbios metabólicos como a doença de Danon, que pode mimetizar a CMH, devem ser considerados.
Função renal: insuficiência renal pode ser observada com disfunção grave do VE.
Fragmento N-terminal do peptídeo natriurético tipo B (NT-proPNB): níveis elevados estão associados a eventos cardiovasculares, insuficiência cardíaca e morte, e podem ter valor diagnóstico, prognóstico e para monitoramento terapêutico.
Troponina: níveis mais elevados estão associados a maior risco de eventos cardiovasculares, insuficiência cardíaca e morte, podendo ter valor diagnóstico, prognóstico e para monitoramento terapêutico.
Urinálise; a proteinúria é sugestiva de comprometimento renal.
Após avaliação especializada, testes adicionais para detectar causas metabólicas e sindrômicas raras são frequentemente necessários em pacientes com cardiomiopatia e características extracardíacas.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Radiografia torácica
A radiografia torácica pode mostrar cardiomegalia secundária à HVE ou aumento do átrio esquerdo, ou pode ser normal.[4]Wigle ED, Rakowski H, Kimball BP, et al. Hypertrophic cardiomyopathy: clinical spectrum and treatment. Circulation. 1995 Oct 1;92(7):1680-92.
https://www.ahajournals.org/doi/10.1161/01.CIR.92.7.1680
http://www.ncbi.nlm.nih.gov/pubmed/7671349?tool=bestpractice.com
Esse exame não é particularmente sensível.[Figure caption and citation for the preceding image starts]: Radiografia torácica de paciente com cardiomiopatia hipertrófica (CMH) demonstrando cardiomegaliaDo acervo de Melanie Everitt, MD, Heart Failure & Transplantation Program, Primary Children's Medical Center, Salt Lake City, UT; usado com permissão [Citation ends].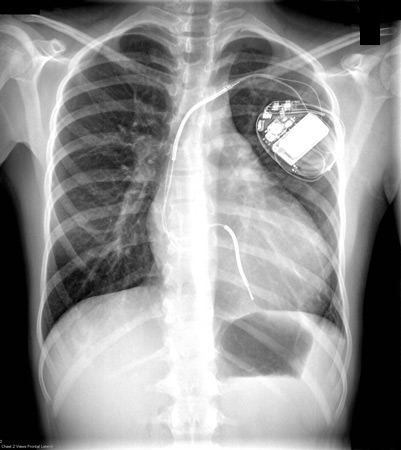
ecocardiografia transtorácica
Na avaliação inicial de todos os pacientes com CMH, recomenda-se ecocardiografia transtorácica 2D e Doppler. O achado clássico é HVE, tipicamente hipertrofia assimétrica do septo.[4]Wigle ED, Rakowski H, Kimball BP, et al. Hypertrophic cardiomyopathy: clinical spectrum and treatment. Circulation. 1995 Oct 1;92(7):1680-92.
https://www.ahajournals.org/doi/10.1161/01.CIR.92.7.1680
http://www.ncbi.nlm.nih.gov/pubmed/7671349?tool=bestpractice.com
[27]Nagueh SF, Phelan D, Abraham T, et al. Recommendations for multimodality cardiovascular imaging of patients with hypertrophic cardiomyopathy: an update from the American Society of Echocardiography, in collaboration with the American Society of Nuclear Cardiology, the Society for Cardiovascular Magnetic Resonance, and the Society of Cardiovascular Computed Tomography. J Am Soc Echocardiogr. 2022 Jun;35(6):533-69.
https://www.doi.org/10.1016/j.echo.2022.03.012
http://www.ncbi.nlm.nih.gov/pubmed/35659037?tool=bestpractice.com
A ecocardiografia também é usada para rastreamento familiar de uma pessoa afetada e para avaliação de risco de morte súbita cardíaca (MSC) em pacientes com diagnóstico conhecido de CMH.
O diagnóstico clínico de CMH é confirmado quando a espessura máxima da parede diastólica final ≥15 mm é obtida em qualquer parte do ventrículo esquerdo (VE). Hipertrofia mais limitada (≥13 mm) pode ser diagnóstica quando presente em familiares de um paciente com CMH ou em conjunto com um teste genético positivo.[2]Writing Committee Members, Ommen SR, Ho CY, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy: a report of the American Heart Association/American College of Cardiology Joint Committee on clinical practice guidelines. J Am Coll Cardiol. 2024 Jun 11;83(23):2324-405.
https://www.sciencedirect.com/science/article/pii/S0735109724003826?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/38727647?tool=bestpractice.com
[27]Nagueh SF, Phelan D, Abraham T, et al. Recommendations for multimodality cardiovascular imaging of patients with hypertrophic cardiomyopathy: an update from the American Society of Echocardiography, in collaboration with the American Society of Nuclear Cardiology, the Society for Cardiovascular Magnetic Resonance, and the Society of Cardiovascular Computed Tomography. J Am Soc Echocardiogr. 2022 Jun;35(6):533-69.
https://www.doi.org/10.1016/j.echo.2022.03.012
http://www.ncbi.nlm.nih.gov/pubmed/35659037?tool=bestpractice.com
O movimento anterior sistólico da valva mitral também pode ser observado, juntamente com a insuficiência mitral.[2]Writing Committee Members, Ommen SR, Ho CY, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy: a report of the American Heart Association/American College of Cardiology Joint Committee on clinical practice guidelines. J Am Coll Cardiol. 2024 Jun 11;83(23):2324-405.
https://www.sciencedirect.com/science/article/pii/S0735109724003826?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/38727647?tool=bestpractice.com
Obstrução da via de saída do VE pode estar presente. Por convenção, a obstrução da via de saída do VE é definida como um pico de gradiente de via de saída do VE no Doppler instantâneo de ≥30 mmHg, mas o limiar para tratamento invasivo é geralmente considerado ≥50 mmHg.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Pode haver anormalidades da função diastólica.[2]Writing Committee Members, Ommen SR, Ho CY, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy: a report of the American Heart Association/American College of Cardiology Joint Committee on clinical practice guidelines. J Am Coll Cardiol. 2024 Jun 11;83(23):2324-405.
https://www.sciencedirect.com/science/article/pii/S0735109724003826?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/38727647?tool=bestpractice.com
A disfunção diastólica deve ser avaliada por Doppler tecidual como parte do exame de rastreamento ecocardiográfico dos parentes de primeiro grau, pois essa anormalidade pode preceder o início de HVE evidente.[27]Nagueh SF, Phelan D, Abraham T, et al. Recommendations for multimodality cardiovascular imaging of patients with hypertrophic cardiomyopathy: an update from the American Society of Echocardiography, in collaboration with the American Society of Nuclear Cardiology, the Society for Cardiovascular Magnetic Resonance, and the Society of Cardiovascular Computed Tomography. J Am Soc Echocardiogr. 2022 Jun;35(6):533-69.
https://www.doi.org/10.1016/j.echo.2022.03.012
http://www.ncbi.nlm.nih.gov/pubmed/35659037?tool=bestpractice.com
[28]Ho CY, Sweitzer NK, McDonough B, et al. Assessment of diastolic function with Doppler tissue imaging to predict genotype in preclinical hypertrophic cardiomyopathy. Circulation. 2002 Jun 25;105(25):2992-7.
https://www.ahajournals.org/doi/10.1161/01.CIR.0000019070.70491.6D
http://www.ncbi.nlm.nih.gov/pubmed/12081993?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: Imagem apical de 4 câmaras demonstrando hipertrofia do septo interventricularDo acervo do Dr Anji T. Yetman, MD, University of Utah; usado com permissão [Citation ends].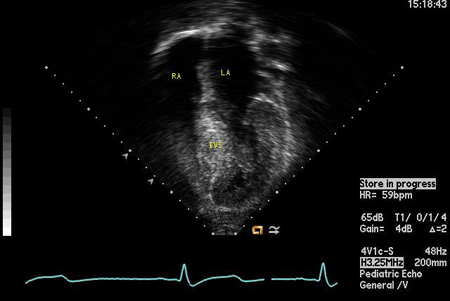 [Figure caption and citation for the preceding image starts]: Visualização de ecocardiografia de eixo longo - hipertrofia septal assimétricaDo acervo do Dr Anji T. Yetman, MD, University of Utah; usado com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Visualização de ecocardiografia de eixo longo - hipertrofia septal assimétricaDo acervo do Dr Anji T. Yetman, MD, University of Utah; usado com permissão [Citation ends].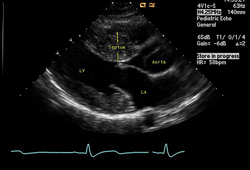
Outros tipos de ecocardiografia
A ecocardiografia sob estresse pode ser útil em pacientes selecionados para avaliar a isquemia miocárdica.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
A ecocardiografia de esforço é útil para identificar a obstrução da via de saída do VE provocada e a regurgitação mitral induzida por exercício em pacientes sintomáticos com CMH.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
A ecocardiografia transesofágica limita-se a indicações selecionadas, como a exclusão de trombos atriais relacionados à fibrilação atrial, a investigação do método de obstrução em pacientes com obstrução da via de saída do VE onde isso não é óbvio, a elucidação do mecanismo da regurgitação mitral ou o planejamento de intervenções invasivas, como miectomia septal.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Eletrocardiograma de esforço
O teste ergométrico é realizado para auxiliar na estratificação de risco. As anormalidades associadas ao aumento do risco de MSC incluem resposta anormal da PA sistólica atenuada de <20 mmHg ao exercício físico, arritmias ventriculares, infradesnivelamento progressivo do segmento ST e sintomas.[29]Frenneaux MP. Assessing the risk of sudden cardiac death in a patient with hypertrophic cardiomyopathy. Heart. 2004 May;90(5):570-5.
https://heart.bmj.com/content/90/5/570
http://www.ncbi.nlm.nih.gov/pubmed/15084566?tool=bestpractice.com
[30]Yetman AT, McCrindle BW, MacDonald C, et al. Myocardial bridging in children with hypertrophic cardiomyopathy - a risk factor for sudden death. N Engl J Med. 1998 Oct 22;339(17):1201-9.
https://www.nejm.org/doi/full/10.1056/NEJM199810223391704
http://www.ncbi.nlm.nih.gov/pubmed/9780340?tool=bestpractice.com
monitoramento com Holter
Isso pode ser normal ou demonstrar arritmias supraventriculares ou ventriculares. Arritmias ventriculares estão associadas a um aumento do risco de morte súbita.[25]Veselka J, Anavekar NS, Charron P. Hypertrophic obstructive cardiomyopathy. Lancet. 2017 Mar 25;389(10075):1253-67.
http://www.ncbi.nlm.nih.gov/pubmed/27912983?tool=bestpractice.com
Técnicas de medicina nuclear
Os pacientes com dor torácica por esforço ou taquicardia ventricular no monitoramento com Holter devem ser submetidos a testes nucleares com tomografia computadorizada por emissão de fóton único ou tomografia por emissão de pósitrons.[27]Nagueh SF, Phelan D, Abraham T, et al. Recommendations for multimodality cardiovascular imaging of patients with hypertrophic cardiomyopathy: an update from the American Society of Echocardiography, in collaboration with the American Society of Nuclear Cardiology, the Society for Cardiovascular Magnetic Resonance, and the Society of Cardiovascular Computed Tomography. J Am Soc Echocardiogr. 2022 Jun;35(6):533-69.
https://www.doi.org/10.1016/j.echo.2022.03.012
http://www.ncbi.nlm.nih.gov/pubmed/35659037?tool=bestpractice.com
A cintilografia de perfusão miocárdica pode demonstrar defeitos de perfusão mesmo na ausência de lesões obstrutivas. Os pacientes podem ter defeitos fixos ou reversíveis. Pacientes com defeitos reversíveis devem ser submetidos a cateterismo cardíaco para a identificação das possíveis causas da isquemia.
A medicina nuclear também pode desempenhar um papel no diagnóstico; é particularmente útil no diagnóstico etiológico da amiloidose cardíaca.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
ressonância nuclear magnética cardíaca (RNMC)
A RNMC com contraste pode ser um complemento útil em pacientes com CMH na avaliação inicial.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
[2]Writing Committee Members, Ommen SR, Ho CY, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy: a report of the American Heart Association/American College of Cardiology Joint Committee on clinical practice guidelines. J Am Coll Cardiol. 2024 Jun 11;83(23):2324-405.
https://www.sciencedirect.com/science/article/pii/S0735109724003826?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/38727647?tool=bestpractice.com
Ela pode auxiliar no diagnóstico e contribuir para a estratificação de risco e o manejo.
A espessura da parede do VE pode ser avaliada; o uso da RNMC pode, assim, aumentar o rendimento diagnóstico em pacientes com suspeita de CMH que apresentam má visualização pela ecocardiografia das paredes ou ápice do VE.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
[2]Writing Committee Members, Ommen SR, Ho CY, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy: a report of the American Heart Association/American College of Cardiology Joint Committee on clinical practice guidelines. J Am Coll Cardiol. 2024 Jun 11;83(23):2324-405.
https://www.sciencedirect.com/science/article/pii/S0735109724003826?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/38727647?tool=bestpractice.com
[27]Nagueh SF, Phelan D, Abraham T, et al. Recommendations for multimodality cardiovascular imaging of patients with hypertrophic cardiomyopathy: an update from the American Society of Echocardiography, in collaboration with the American Society of Nuclear Cardiology, the Society for Cardiovascular Magnetic Resonance, and the Society of Cardiovascular Computed Tomography. J Am Soc Echocardiogr. 2022 Jun;35(6):533-69.
https://www.doi.org/10.1016/j.echo.2022.03.012
http://www.ncbi.nlm.nih.gov/pubmed/35659037?tool=bestpractice.com
A função sistólica e diastólica também pode ser avaliada, bem como a função da valva mitral, a obstrução da via de saída do VE e as dimensões do átrio esquerdo.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
O uso de técnicas de realce tardio com gadolínio pode identificar áreas da fibrose miocárdica, podendo ser um marcador para desfechos adversos ou auxiliando na diferenciação da CMH do coração de atleta.[31]O’Hanlon R, Assomull RG, Prasad SK. Use of cardiovascular magnetic resonance for diagnosis and management in hypertrophic cardiomyopathy. Curr Cardiol Rep. 2007 Mar;9(1):51-6.
http://www.ncbi.nlm.nih.gov/pubmed/17362685?tool=bestpractice.com
A RNMC está surgindo como forma de identificar pacientes com aumento do risco de arritmias. Vários estudos consideraram a presença de fibrose miocárdica por realce tardio com gadolínio associada à ocorrência de arritmias ventriculares, bem como um fator de risco independente para morte.[2]Writing Committee Members, Ommen SR, Ho CY, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy: a report of the American Heart Association/American College of Cardiology Joint Committee on clinical practice guidelines. J Am Coll Cardiol. 2024 Jun 11;83(23):2324-405.
https://www.sciencedirect.com/science/article/pii/S0735109724003826?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/38727647?tool=bestpractice.com
[32]Bruder O, Wagner A, Jensen CJ, et al. Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2010 Sep 7;56(11):875-87.
https://www.jacc.org/doi/10.1016/j.jacc.2010.05.007
http://www.ncbi.nlm.nih.gov/pubmed/20667520?tool=bestpractice.com
[33]Adabag AS, Maron BJ, Appelbaum E, et al. Occurrence and frequency of arrythmias in hypertrophic cardiomyopathy in relation to delayed enhancement on cardiovascular magnetic resonance. J Am Coll Cardiol. 2008 Apr 8;51(14):1369-74.
http://www.ncbi.nlm.nih.gov/pubmed/18387438?tool=bestpractice.com
[34]Fluechter S, Kuschyk J, Wolpert C, et al. Extent of late gadolinium enhancement detected by cardiovascular magnetic resonance correlates with the inducibility of ventricular tachyarrhythmia in hypertrophic cardiomyopathy. J Cardiovasc Magn Reson. 2010 May 21;12:30.
https://jcmr-online.biomedcentral.com/articles/10.1186/1532-429X-12-30
http://www.ncbi.nlm.nih.gov/pubmed/20492668?tool=bestpractice.com
[35]Suk T, Edwards C, Hart H, et al. Myocardial scar detected by contrast-enhanced cardiac magnetic resonance imaging is associated with ventricular tachycardia in hypertrophic cardiomyopathy patients. Heart Lung Circ. 2008 Oct;17(5):370-4.
http://www.ncbi.nlm.nih.gov/pubmed/18562248?tool=bestpractice.com
[36]Leonardi S, Raineri C, De Ferrari GM, et al. Usefulness of cardiac magnetic resonance in assessing the risk of ventricular arrhythmias and sudden death in patients with hypertrophic cardiomyopathy. Eur Heart J. 2009 Aug;30(16):2003-10.
https://academic.oup.com/eurheartj/article/30/16/2003/630669
http://www.ncbi.nlm.nih.gov/pubmed/19474054?tool=bestpractice.com
[37]O'Hanlon R, Grasso A, Roughton M, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2010 Sep 7;56(11):867-74.
https://www.jacc.org/doi/10.1016/j.jacc.2010.05.010
http://www.ncbi.nlm.nih.gov/pubmed/20688032?tool=bestpractice.com
A caracterização tecidual na RNMC pode fornecer pistas sobre a etiologia, uma vez que achados característicos estão associados a certas doenças, por exemplo, na CMH sarcomérica, é típica uma parede média irregular em áreas hipertrofiadas, enquanto na hipertrofia cardíaca relacionada à amiloidose, realce tardio com gadolínio subendocárdico difuso é observado. Esses achados devem ser avaliados coletivamente com resultados genéticos e outras características clínicas por operadores especialistas em imagens cardíacas e na avaliação de doenças do músculo cardíaco.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
A RNMC de acompanhamento em série, a cada 2-5 anos, dependendo da gravidade inicial e da evolução clínica, pode ajudar na avaliação da progressão da doença, bem como dos benefícios da terapia.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Tomografia computadorizada cardíaca (TC)
Embora não seja comumente usada, a TC pode fornecer informações importantes quando a ecocardiografia é tecnicamente limitada e a RNMC é contraindicada ou indisponível.[2]Writing Committee Members, Ommen SR, Ho CY, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy: a report of the American Heart Association/American College of Cardiology Joint Committee on clinical practice guidelines. J Am Coll Cardiol. 2024 Jun 11;83(23):2324-405.
https://www.sciencedirect.com/science/article/pii/S0735109724003826?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/38727647?tool=bestpractice.com
A TC cardíaca fornece uma definição clara da estrutura do VE (incluindo padrão de hipertrofia, medição da espessura da parede, detecção de membrana subaórtica e trombo intracardíaco) e função. As desvantagens da TC são o uso de radiação e contraste de radioiodo e resolução temporal inferior em comparação com a ecocardiografia.[2]Writing Committee Members, Ommen SR, Ho CY, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy: a report of the American Heart Association/American College of Cardiology Joint Committee on clinical practice guidelines. J Am Coll Cardiol. 2024 Jun 11;83(23):2324-405.
https://www.sciencedirect.com/science/article/pii/S0735109724003826?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/38727647?tool=bestpractice.com
angiografia coronariana por tomografia computadorizada
Indicada em pacientes com dor torácica por esforço ou isquemia em testes nucleares para verificar a presença de doença arterial coronariana concomitante ou ponte miocárdica, comorbidades que podem afetar as manifestações clínicas e a evolução da CMH.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
[2]Writing Committee Members, Ommen SR, Ho CY, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy: a report of the American Heart Association/American College of Cardiology Joint Committee on clinical practice guidelines. J Am Coll Cardiol. 2024 Jun 11;83(23):2324-405.
https://www.sciencedirect.com/science/article/pii/S0735109724003826?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/38727647?tool=bestpractice.com
[27]Nagueh SF, Phelan D, Abraham T, et al. Recommendations for multimodality cardiovascular imaging of patients with hypertrophic cardiomyopathy: an update from the American Society of Echocardiography, in collaboration with the American Society of Nuclear Cardiology, the Society for Cardiovascular Magnetic Resonance, and the Society of Cardiovascular Computed Tomography. J Am Soc Echocardiogr. 2022 Jun;35(6):533-69.
https://www.doi.org/10.1016/j.echo.2022.03.012
http://www.ncbi.nlm.nih.gov/pubmed/35659037?tool=bestpractice.com
Cateterismo cardíaco
Em pacientes sintomáticos com CMH e imagens cardíacas não invasivas inconclusivas, o cateterismo cardíaco esquerdo e direito pode ser considerado para avaliar a gravidade da obstrução da via de saída do VE e para medir as pressões de enchimento do VE.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
[2]Writing Committee Members, Ommen SR, Ho CY, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy: a report of the American Heart Association/American College of Cardiology Joint Committee on clinical practice guidelines. J Am Coll Cardiol. 2024 Jun 11;83(23):2324-405.
https://www.sciencedirect.com/science/article/pii/S0735109724003826?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/38727647?tool=bestpractice.com
Também pode ser usado para verificar a presença de doença coronariana ou ponte miocárdica concomitantes, e é recomendado para os pacientes candidatos à terapia de redução septal.[2]Writing Committee Members, Ommen SR, Ho CY, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy: a report of the American Heart Association/American College of Cardiology Joint Committee on clinical practice guidelines. J Am Coll Cardiol. 2024 Jun 11;83(23):2324-405.
https://www.sciencedirect.com/science/article/pii/S0735109724003826?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/38727647?tool=bestpractice.com
Biópsia endomiocárdica
A biópsia endomiocárdica geralmente não é recomendada para o diagnóstico de CMH, mas pode ser considerada em raras ocasiões, especialmente quando o padrão de hipertrofia é difuso e há suspeita de outras cardiomiopatias que apresentam hipertrofia.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Estratificação de risco
Risco de morte súbita cardíaca (MSC)
Após o diagnóstico, os pacientes devem ser submetidos à estratificação de risco, incluindo o monitoramento por Holter e o eletrocardiograma de esforço (a menos que seja contraindicado) para definir melhor o risco de morte súbita.[2]Writing Committee Members, Ommen SR, Ho CY, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy: a report of the American Heart Association/American College of Cardiology Joint Committee on clinical practice guidelines. J Am Coll Cardiol. 2024 Jun 11;83(23):2324-405.
https://www.sciencedirect.com/science/article/pii/S0735109724003826?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/38727647?tool=bestpractice.com
A European Society of Cardiology (ESC) desenvolveu uma calculadora de predição do risco para MSC em 5 anos em pacientes ≥16 anos com CMH.[38]O'Mahony C, Jichi F, Pavlou M, et al. A novel clinical risk prediction model for sudden cardiac death in hypertrophic cardiomyopathy (HCM risk-SCD). Eur Heart J. 2014 Aug 7;35(30):2010-20.
https://academic.oup.com/eurheartj/article/35/30/2010/467191
http://www.ncbi.nlm.nih.gov/pubmed/24126876?tool=bestpractice.com
O cálculo do risco é baseado em idade, espessura máxima da parede VE, diâmetro do átrio esquerdo, gradiente da via de saída do VE, história familiar de MSC, taquicardia ventricular não sustentada e síncope inexplicada. O risco de MSC deve ser reavaliado em intervalos de 1-2 anos ou sempre que houver alteração do estado clínico.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Esses modelos não devem ser utilizados em atletas de elite ou em indivíduos com doenças metabólicas/infiltrantes (por exemplo, doença de Anderson-Fabry) e síndromes (por exemplo, síndrome de Noonan).[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Deve-se observar que os modelos atuais de estratificação do; risco podem não ser confiáveis na predição de uma morte súbita futura, e a colocação de um cardioversor-desfibrilador implantável (CDI) ainda pode ser justificada em pacientes com escores de baixo risco.[39]Maron BJ, Casey SA, Chan RH, et al. Independent assessment of the European Society of Cardiology sudden death risk model for hypertrophic cardiomyopathy. Am J Cardiol. 2015 Sep 1;116(5):757-64.
http://www.ncbi.nlm.nih.gov/pubmed/26183790?tool=bestpractice.com
As diretrizes dos EUA refletem essa incerteza, recomendando que a ferramenta seja usada como um auxílio ao processo de tomada de decisão compartilhada para a colocação de CDI em pacientes com marcadores de risco clínico, enquanto as diretrizes europeias recomendam que ela seja usada como base para a tomada de decisão de todos os pacientes com CMH.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
[2]Writing Committee Members, Ommen SR, Ho CY, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy: a report of the American Heart Association/American College of Cardiology Joint Committee on clinical practice guidelines. J Am Coll Cardiol. 2024 Jun 11;83(23):2324-405.
https://www.sciencedirect.com/science/article/pii/S0735109724003826?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/38727647?tool=bestpractice.com
As diretrizes da ESC recomendam que a implantação de um CDI seja considerada em pacientes com risco estimado de morte súbita em 5 anos ≥6%, após avaliação clínica detalhada que considere: (i) o risco de complicações ao longo da vida; (ii) risco competitivo de mortalidade pela doença e comorbidades; E (iii) o impacto de um CDI no estilo de vida, na situação socioeconômica e na saúde psicológica. Também pode ser considerada em pacientes com risco entre ≥4% e <6% individualmente. Para pacientes que estão na categoria de baixo risco (<4% de risco estimado de MSC em 5 anos), o CDI geralmente não é recomendado. No entanto, as diretrizes reconhecem que o CDI pode ser considerado em pacientes de baixo risco que apresentam realce tardio com gadolínio extenso (≥15%) na RNMC ou fração de ejeção do VE <50%; esses fatores não fazem parte do risco de MSC para CMH, mas as evidências sugerem que aumentam o risco de MSC. Recomenda-se a tomada de decisão compartilhada.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Os fatores de risco para MSC são os seguintes:[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
[2]Writing Committee Members, Ommen SR, Ho CY, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy: a report of the American Heart Association/American College of Cardiology Joint Committee on clinical practice guidelines. J Am Coll Cardiol. 2024 Jun 11;83(23):2324-405.
https://www.sciencedirect.com/science/article/pii/S0735109724003826?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/38727647?tool=bestpractice.com
[40]Goldberger JJ, Cain ME, Hohnloser SH, et al. American Heart Association/American College of Cardiology Foundation/Heart Rhythm Society scientific statement on noninvasive risk stratification techniques for identifying patients at risk for sudden cardiac death. J Am Coll Cardiol. 2008 Sep 30;52(14):1179-99.
https://www.jacc.org/doi/10.1016/j.jacc.2008.05.003
http://www.ncbi.nlm.nih.gov/pubmed/18804749?tool=bestpractice.com
Idade mais jovem: alguns estudos relataram um aumento do risco significativo de MSC em pacientes mais jovens.
Taquicardia ventricular não sustentada (definida como ≥3 batimentos ventriculares consecutivos a ≥120 batimentos por minuto com duração <30 segundos) no monitor Holter: ocorre em 20% a 30% dos pacientes.
Resposta anormal da pressão arterial (PA) à atividade física: definida como um aumento na PA sistólica de <20 mmHg, uma ausência de aumento ou uma queda na PA de >20 mmHg durante o exercício. Medicamentos podem afetar a resposta da PA, e devem ser levados em conta na interpretação dos resultados do teste ergométrico.
Hipertrofia maciça (espessura da parede ventricular esquerda [VE] ≥30 mm).
Obstrução da via de saída do VE grave por ecocardiografia (obstrução da via de saída do VE >30 a 50 mmHg): embora a obstrução grave seja considerada um fator de risco menor para morte súbita, o grau de obstrução da via de saída geralmente não se correlaciona com o risco de morte súbita. A terapia medicamentosa ou a cirurgia para reduzir a obstrução da via de saída não diminui o risco de morte súbita.
História familiar de morte súbita: embora as definições variem, uma história familiar de MSC é geralmente considerada clinicamente significativa quando um ou mais parentes de primeiro grau morreram repentinamente com idade <40 anos, com ou sem diagnóstico de CMH, ou quando a MSC ocorreu em um parente de primeiro grau em qualquer idade com diagnóstico estabelecido de CMH.
História pessoal de síncope inexplicada.
Parada cardíaca prévia ou taquicardia ventricular sustentada.
Disfunção sistólica do VE com fração de ejeção <50%.
Aumento do átrio esquerdo.
Presença de aneurisma apical do VE.
Realce tardio com gadolínio difuso e extenso por RNM cardíaca: a ressonância nuclear magnética cardíaca está emergindo como um meio de identificar pacientes com aumento do risco de arritmias. Inúmeros estudos revelaram que a presença da fibrose miocárdica por realce tardio com gadolínio está associada à ocorrência de arritmias ventriculares.[33]Adabag AS, Maron BJ, Appelbaum E, et al. Occurrence and frequency of arrythmias in hypertrophic cardiomyopathy in relation to delayed enhancement on cardiovascular magnetic resonance. J Am Coll Cardiol. 2008 Apr 8;51(14):1369-74.
http://www.ncbi.nlm.nih.gov/pubmed/18387438?tool=bestpractice.com
[34]Fluechter S, Kuschyk J, Wolpert C, et al. Extent of late gadolinium enhancement detected by cardiovascular magnetic resonance correlates with the inducibility of ventricular tachyarrhythmia in hypertrophic cardiomyopathy. J Cardiovasc Magn Reson. 2010 May 21;12:30.
https://jcmr-online.biomedcentral.com/articles/10.1186/1532-429X-12-30
http://www.ncbi.nlm.nih.gov/pubmed/20492668?tool=bestpractice.com
[35]Suk T, Edwards C, Hart H, et al. Myocardial scar detected by contrast-enhanced cardiac magnetic resonance imaging is associated with ventricular tachycardia in hypertrophic cardiomyopathy patients. Heart Lung Circ. 2008 Oct;17(5):370-4.
http://www.ncbi.nlm.nih.gov/pubmed/18562248?tool=bestpractice.com
[36]Leonardi S, Raineri C, De Ferrari GM, et al. Usefulness of cardiac magnetic resonance in assessing the risk of ventricular arrhythmias and sudden death in patients with hypertrophic cardiomyopathy. Eur Heart J. 2009 Aug;30(16):2003-10.
https://academic.oup.com/eurheartj/article/30/16/2003/630669
http://www.ncbi.nlm.nih.gov/pubmed/19474054?tool=bestpractice.com
[37]O'Hanlon R, Grasso A, Roughton M, et al. Prognostic significance of myocardial fibrosis in hypertrophic cardiomyopathy. J Am Coll Cardiol. 2010 Sep 7;56(11):867-74.
https://www.jacc.org/doi/10.1016/j.jacc.2010.05.010
http://www.ncbi.nlm.nih.gov/pubmed/20688032?tool=bestpractice.com
A presença de fibrose também foi relatada como um risco independente para a morte.[32]Bruder O, Wagner A, Jensen CJ, et al. Myocardial scar visualized by cardiovascular magnetic resonance imaging predicts major adverse events in patients with hypertrophic cardiomyopathy. J Am Coll Cardiol. 2010 Sep 7;56(11):875-87.
https://www.jacc.org/doi/10.1016/j.jacc.2010.05.007
http://www.ncbi.nlm.nih.gov/pubmed/20667520?tool=bestpractice.com
Considerações para comorbidades
Isquemia
A isquemia pode estar relacionada a uma ponte miocárdica, obstrução da via de saída do VE ou hipertrofia maciça com redução da perfusão miocárdica. A presença de isquemia é um fator de risco fraco para MSC. Os pacientes com angina ou infradesnivelamento do segmento ST ao eletrocardiograma de esforço devem ser avaliados para isquemia com uma cintilografia ou angiotomografia se a probabilidade de doença arterial coronariana (DAC) for relativamente baixa. Uma angiotomografia ou cateterismo cardíaco são indicados se houver maior probabilidade de DAC considerando-se outros fatores do paciente.[2]Writing Committee Members, Ommen SR, Ho CY, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy: a report of the American Heart Association/American College of Cardiology Joint Committee on clinical practice guidelines. J Am Coll Cardiol. 2024 Jun 11;83(23):2324-405.
https://www.sciencedirect.com/science/article/pii/S0735109724003826?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/38727647?tool=bestpractice.com
[41]Lubarsky L, Gupta MP, Hecht HS. Evaluation of myocardial bridging of the left anterior descending coronary artery by 64-slice multidetector computed tomographic angiography. Am J Cardiol. 2007 Oct 1;100(7):1081-2.
http://www.ncbi.nlm.nih.gov/pubmed/17884365?tool=bestpractice.com
Uma ponte miocárdica (tunelização das artérias coronárias no músculo cardíaco) também deve ser considerada no cenário de uma angina ou isquemia. O cateterismo cardíaco ou a angiotomografia podem ser usados para avaliar a formação de ponte, e o avaliador deve dar atenção específica a esse possível diagnóstico.[42]Kantarci M, Doganay S, Karcaaltincaba M, et al. Clinical situations in which coronary CT angiography confers superior diagnostic information compared with coronary angiography. Diagn Interv Radiol. 2012 May-Jun;18(3):261-9.
https://www.dirjournal.org/en/clinical-situations-in-which-coronary-ct-angiography-confers-superior-diagnostic-information-compared-with-coronary-angiography-166357
http://www.ncbi.nlm.nih.gov/pubmed/22261852?tool=bestpractice.com
Insuficiência cardíaca
Os pacientes com CMH podem apresentar sintomas de insuficiência cardíaca (dispneia; tosse persistente; edema nos tornozelos, fadiga).[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
A ecocardiografia transtorácica (que é realizada na avaliação inicial de todos os pacientes com CMH) permite a determinação precisa da função sistólica e diastólica biventricular. A medição do NT-proPNB também é recomendada; níveis elevados estão associados a eventos cardiovasculares, insuficiência cardíaca e morte, e podem ter valor diagnóstico, prognóstico e de monitoramento terapêutico.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Fibrilação atrial (FA)
Nos pacientes com alto risco de FA, o monitoramento ambulatorial prolongado com ECG deve ser considerado à avaliação inicial e como parte do acompanhamento anual.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
[2]Writing Committee Members, Ommen SR, Ho CY, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy: a report of the American Heart Association/American College of Cardiology Joint Committee on clinical practice guidelines. J Am Coll Cardiol. 2024 Jun 11;83(23):2324-405.
https://www.sciencedirect.com/science/article/pii/S0735109724003826?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/38727647?tool=bestpractice.com
Análise de mutação genética
Os testes genéticos em um indivíduo com cardiomiopatia (conhecidos como testes confirmatórios ou testes de diagnóstico) são recomendados pelo seu benefício direto: (i) para confirmar o diagnóstico; (ii) onde pode informar o prognóstico; (iii) onde pode informar a seleção do tratamento; ou (iv) onde possa informar seu manejo reprodutivo. Os testes genéticos de um indivíduo afetado também podem ser indicados se houver familiares que possam se beneficiar dos testes, particularmente aqueles que serão incluídos na vigilância em longo prazo se a etiologia genética não for estabelecida (e que podem ser poupados deste fardo se um diagnóstico genético for realizado na família).[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
A utilidade clínica do teste genético tem limitações. Acredita-se que os genes causadores da doença identificados atualmente sejam responsáveis por apenas 80% dos casos. Além disso, a sensibilidade do teste genético disponível comercialmente depende do número de genes rastreados pelo laboratório em particular, podendo ser <80%. Quando as 8 mutações mais comuns do sarcômero são rastreadas, a sensibilidade clínica aproxima-se de 60%.[17]Alcalai R, Seidman JG, Seidman CE. Genetic basis of hypertrophic cardiomyopathy: from bench to the clinics. J Cardiovasc Electrophysiol. 2008 Jan;19(1):104-10.
https://onlinelibrary.wiley.com/doi/10.1111/j.1540-8167.2007.00965.x
http://www.ncbi.nlm.nih.gov/pubmed/17916152?tool=bestpractice.com
Em até 40% dos pacientes com CMH, nenhuma variante do sarcômero é identificada e não há história familiar da doença.[43]Wilde AAM, Semsarian C, Márquez MF, et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the state of genetic testing for cardiac diseases. Europace. 2022 Sep 1;24(8):1307-67.
https://academic.oup.com/europace/article/24/8/1307/6562982?login=false
http://www.ncbi.nlm.nih.gov/pubmed/35373836?tool=bestpractice.com
A ausência de uma variante monogênica causadora de doença nos testes genéticos convencionais deixa três possibilidades: (i) existe uma causa monogênica que não foi identificada (ou seja, não detectada ou reconhecida como causadora pelos testes atuais); (ii) a cardiomiopatia não tem etiologia genética; ou (iii) a cardiomiopatia é atribuível aos efeitos de múltiplas variantes de efeito individualmente menor.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
Apesar das suas limitações, o teste genético é importante no rastreamento dos familiares de um paciente afetado com uma mutação identificada. Os testes genéticos nessa situação determinarão quem requer avaliação clínica contínua (conhecido como teste em cascata):[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
[2]Writing Committee Members, Ommen SR, Ho CY, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy: a report of the American Heart Association/American College of Cardiology Joint Committee on clinical practice guidelines. J Am Coll Cardiol. 2024 Jun 11;83(23):2324-405.
https://www.sciencedirect.com/science/article/pii/S0735109724003826?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/38727647?tool=bestpractice.com
[43]Wilde AAM, Semsarian C, Márquez MF, et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the state of genetic testing for cardiac diseases. Europace. 2022 Sep 1;24(8):1307-67.
https://academic.oup.com/europace/article/24/8/1307/6562982?login=false
http://www.ncbi.nlm.nih.gov/pubmed/35373836?tool=bestpractice.com
[44]Musunuru K, Hershberger RE, Day SM, et al. Genetic testing for inherited cardiovascular diseases: a scientific statement from the American Heart Association. Circ Genom Precis Med. 2020 Aug;13(4):e000067.
https://www.ahajournals.org/doi/10.1161/HCG.0000000000000067
http://www.ncbi.nlm.nih.gov/pubmed/32698598?tool=bestpractice.com
Parentes com mutação identificada devem continuar o rastreamento para o desenvolvimento clínico de CMH. O desenvolvimento de doença clinicamente aparente pode ocorrer tardiamente na idade adulta, então o rastreamento deverá ser vitalício.
Os parentes negativos para o gene podem ser assegurados de que não têm a mutação causadora da doença e de que não necessitam de rastreamento adicional.
Embora a identificação de uma mutação genética durante os testes em cascata indique que o desenvolvimento de CMH é muito provável, existe variabilidade genótipo-fenótipo. Apesar das mutações genéticas idênticas, a mutação genética pode manifestar-se como CMH, cardiomiopatia restritiva, cardiomiopatia dilatada ou nenhuma anormalidade clinicamente aparente em diferentes pacientes. Para aqueles sem a doença visível, a penetrância de início tardio deve ser considerada.[45]Christiaans I, Birnie E, van Langen IM, et al. The yield of risk stratification for sudden cardiac death in hypertrophic cardiomyopathy myosin-binding protein C gene mutation carriers: focus on predictive screening. Eur Heart J. 2010 Apr;31(7):842-8.
https://academic.oup.com/eurheartj/article/31/7/842/431846
http://www.ncbi.nlm.nih.gov/pubmed/20019025?tool=bestpractice.com
O risco de morte súbita pode ser menor ou maior para a mesma mutação.[46]Ho CY. Genetics and clinical destiny: improving care in hypertrophic cardiomyopathy. Circulation. 2010 Dec 7;122(23):2430-40.
https://www.ahajournals.org/doi/10.1161/CIRCULATIONAHA.110.978924
http://www.ncbi.nlm.nih.gov/pubmed/21135371?tool=bestpractice.com
O aconselhamento genético deve estar disponível para todos os pacientes aos quais são oferecidos testes genéticos, para informar a tomada de decisões e garantir que os resultados possam ser revistos e o seu significado clínico determinado de forma adequada.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
A importância das potenciais implicações psicológicas, sociais, legais, éticas e profissionais de ter uma doença genética também deve ser discutida e deve ser fornecido apoio adequado.[2]Writing Committee Members, Ommen SR, Ho CY, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy: a report of the American Heart Association/American College of Cardiology Joint Committee on clinical practice guidelines. J Am Coll Cardiol. 2024 Jun 11;83(23):2324-405.
https://www.sciencedirect.com/science/article/pii/S0735109724003826?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/38727647?tool=bestpractice.com
O aconselhamento genético pré-natal deve ser oferecido aos pais que tiveram um filho previamente afetado com CMH hereditária devido a uma ou mais variantes patogênicas, ou a casais onde um ou ambos os parceiros são portadores de uma variante patogênica conhecida.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
[47]Joglar JA, Kapa S, Saarel EV, et al. 2023 HRS expert consensus statement on the management of arrhythmias during pregnancy. Heart Rhythm. 2023 Oct;20(10):e175-e264.
https://www.heartrhythmjournal.com/article/S1547-5271(23)02246-4/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/37211147?tool=bestpractice.com
O risco de transmissão da doença deve ser discutido, bem como as possíveis opções reprodutivas (por exemplo, fertilização in vitro com diagnóstico genético pré-implantacional, rastreamento genético pré-natal e testes genéticos pós-parto).[2]Writing Committee Members, Ommen SR, Ho CY, et al. 2024 AHA/ACC/AMSSM/HRS/PACES/SCMR guideline for the management of hypertrophic cardiomyopathy: a report of the American Heart Association/American College of Cardiology Joint Committee on clinical practice guidelines. J Am Coll Cardiol. 2024 Jun 11;83(23):2324-405.
https://www.sciencedirect.com/science/article/pii/S0735109724003826?via%3Dihub
http://www.ncbi.nlm.nih.gov/pubmed/38727647?tool=bestpractice.com
[43]Wilde AAM, Semsarian C, Márquez MF, et al. European Heart Rhythm Association (EHRA)/Heart Rhythm Society (HRS)/Asia Pacific Heart Rhythm Society (APHRS)/Latin American Heart Rhythm Society (LAHRS) Expert Consensus Statement on the state of genetic testing for cardiac diseases. Europace. 2022 Sep 1;24(8):1307-67.
https://academic.oup.com/europace/article/24/8/1307/6562982?login=false
http://www.ncbi.nlm.nih.gov/pubmed/35373836?tool=bestpractice.com
Os avanços na tecnologia de sequenciamento genético e o aumento da acessibilidade aos testes levaram a um número crescente de variantes genéticas identificadas incidentalmente associadas à CMH. Interpretar a relevância clínica de tais achados pode ser um desafio; a American Heart Association produziu orientações sobre como manejá-los, com ênfase em uma abordagem de equipe multidisciplinar.[48]Landstrom AP, Chahal AA, Ackerman MJ, et al. Interpreting incidentally identified variants in genes associated with heritable cardiovascular disease: a scientific statement from the American Heart Association. Circ Genom Precis Med. 2023 Apr;16(2):e000092.
https://www.ahajournals.org/doi/full/10.1161/HCG.0000000000000092?rfr_dat=cr_pub++0pubmed&url_ver=Z39.88-2003&rfr_id=ori%3Arid%3Acrossref.org
http://www.ncbi.nlm.nih.gov/pubmed/36970980?tool=bestpractice.com
O American College of Medical Genetics and Genomics recomendou que os genes associados à cardiomiopatia sejam avaliados quanto a achados secundários sempre que um sequenciamento clínico amplo for realizado, independentemente da indicação inicial para o teste.[49]National Library of Medicine. ACMG recommendations for reporting of secondary findings in clinical exome and genome sequencing [internet publication].
https://www.ncbi.nlm.nih.gov/clinvar/docs/acmg
No entanto, atualmente não há consenso internacional em torno dessa recomendação.[1]Arbelo E, Protonotarios A, Gimeno JR, et al. 2023 ESC Guidelines for the management of cardiomyopathies. Eur Heart J. 2023 Oct 1;44(37):3503-626.
https://academic.oup.com/eurheartj/article/44/37/3503/7246608?login=false
http://www.ncbi.nlm.nih.gov/pubmed/37622657?tool=bestpractice.com
