Abordagem
A retinite pigmentosa (RP) é uma doença incurável e permanente que leva à perda progressiva da visão. Devido às implicações de tal diagnóstico, é importante que os pacientes com suspeita de RP sejam atendidos por um especialista. Se a RP for confirmada, as associações sindrômicas devem ser consideradas e totalmente avaliadas.
História
A perda da visão é gradual e progressiva. A acuidade visual varia de uma visão perfeita 20/20 até a mera detecção de luz. É incomum, no entanto, haver perda total da visão, e algum grau de visão é geralmente mantido até uma idade avançada. A acuidade central pode estar diminuída em formas avançadas do tipo de doença mais comum, a distrofia bastonetes-cones, ou em um momento anterior, se o edema macular cistoide se desenvolver.
Os sintomas dependem de qual tipo de fotorreceptores estão afetados. Mais comumente, os bastonetes são afetados primeiro, e alguns dos sintomas precoces incluem a dificuldade em dirigir à noite devido à visão comprometida, e a adaptação prejudicada quando o indivíduo entra em um ambiente escuro. A perda da visão periférica é outra característica, e o paciente pode relatar que esbarra nos móveis, na borda das portas ou que tem dificuldade para praticar esportes com raquete. A perda do campo de visão frequentemente não é notada até que esta atinge um estágio moderado devido à sobreposição dos campos visuais de cada olho.
Na RP do tipo cones-bastonetes, na qual os cones são predominantemente afetados primeiro, os sintomas podem incluir dificuldade em ler ou enxergar detalhes (devido à perda de visão central) ou comprometimento da visão das cores. Os pacientes podem declarar que enxergam flashes de luz ou raios luminosos, embora esses relatos também possam acontecer na enxaqueca com aura ou no descolamento da retina. No entanto, com a RP, os flashes são contínuos em vez de episódicos. O ofuscamento por luzes fortes pode ser um problema na doença avançada.
Um subtipo grave de RP, conhecido como amaurose congênita de Leber, pode se manifestar na primeira infância com visão diminuída, pupilas lentas e nistagmo.[5]
Muitos pacientes com RP ligada ao cromossomo X ou dominante têm história familiar da doença. A herança autossômica recessiva pode ou não revelar uma história familiar. As formas ligadas ao cromossomo X afetam apenas a prole do sexo masculino (embora as mulheres possam ser portadoras); portanto, a história pode não ficar evidente se não houver meninos dentro de uma geração. Geralmente, as formas da doença ligadas ao cromossomo X se manifestam na infância, enquanto as formas autossômicas dominante e recessiva tendem a aparecer mais tarde.[4]
RP sindrômica
A RP é mais frequentemente encontrada isoladamente, mas também pode existir como parte de uma síndrome. Para se descartar a presença de doença sindrômica, é importante procurar outros sintomas e sinais: em particular, problemas de audição ou de equilíbrio (Usher, Alstrom), obesidade (Bardet-Biedl), insuficiência renal (Bardet-Biedl, Alstrom, Senior-Loken), dedos e pododáctilos extras (Bardet-Biedl, Joubert), ataxia (Joubert, Bardet-Biedl), convulsões (lipofuscinose ceroide neuronal) e diabetes mellitus (doença de Alstrom). A perda auditiva, a ataxia, a oftalmoplegia, os defeitos da condução cardíaca e a disfagia podem ser observados na síndrome de Kearns-Sayre. A abetalipoproteinemia é uma causa de RP potencialmente reversível, já que muitas das características ocorrem devido à falta de absorção de vitaminas lipossolúveis. Se diagnosticados precocemente, os problemas retinianos podem ser prevenidos ou retardados por suplementação com vitaminas A e E. A doença de Refsum infantil geralmente se manifesta na primeira infância com comprometimento da audição, problemas de coordenação e tônus muscular diminuído. Na doença de Refsum adulta, as características podem incluir anosmia, ataxia e arritmias cardíacas. O encaminhamento a um geneticista clínico pode ser necessário para avaliação e diagnóstico abrangentes dessas condições sindrômicas.
Exame oftalmológico
O Comitê de Padrão de Prática Preferencial da American Academy of Ophthalmology relatou recomendações para a frequência de exames oftalmológicos abrangentes em adultos para pacientes assintomáticos, e para aqueles pacientes com ou sem fatores de risco para doenças oculares.[30]
Acuidade visual
A acuidade visual pode ser mensurada usando uma tabela de Snellen e pode variar dependendo do tipo e da gravidade da RP. É essencial registrar a acuidade visual para as implicações legais e funcionais e para acompanhar a evolução. Erros de refração podem ser notados e a visão pode ser melhorada através do uso de óculos se esta for corrigida, embora a RP subjacente permaneça. Em formas graves precoces de RP, como a amaurose congênita de Leber, a hiperopia predomina. Em formas de início tardio de RP, a miopia e o astigmatismo são comuns. Cerca de metade dos pacientes com RP desenvolverão cataratas, e sua remoção pode melhorar a visão se a RP subjacente não tiver progredido muito.
Exame do segmento anterior e medição da pressão intraocular
A pressão intraocular de cada olho deve ser medida antes da dilatação, na apresentação e em cada consulta subsequente.
O exame completo da córnea, conjuntiva, segmento anterior e cristalino deve ser realizado na apresentação e em cada consulta. Pacientes com RP tendem a desenvolver catarata mais cedo do que o esperado, particularmente alterações na catarata subcapsular posterior.
Fundoscopia
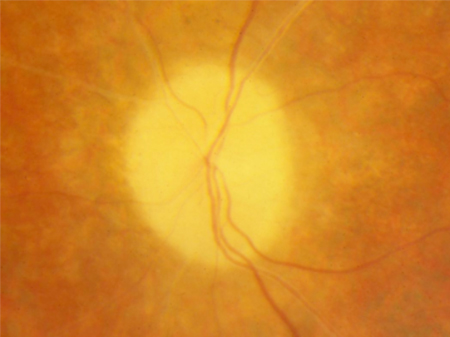
Tipicamente, o disco óptico está pálido e o nervo óptico apresenta uma aparência em cera, embora possa parecer normal no início da doença.
A atenuação vascular dos vasos retinianos ajuda a distinguir a RP da coroideremia (que demonstra vasos retinianos normais, mas atrofia dos vasos coroidais).[Figure caption and citation for the preceding image starts]: Palidez em cera e atenuação vascularDo acervo do Oregon Retinal Degeneration Center [Citation ends].
A degeneração do epitélio pigmentar da retina é a principal característica. A pigmentação em espículas ósseas resulta da migração das células do epitélio pigmentar da retina para dentro da mesma, frequentemente circundando vasos retinianos. A aparência é de pontos ou aglomerados de pontos negros na retina.[Figure caption and citation for the preceding image starts]: Espículas ósseasDo acervo do Oregon Retinal Degeneration Center [Citation ends].
 [Figure caption and citation for the preceding image starts]: Retinite pigmentosaDo acervo do Oregon Retinal Degeneration Center [Citation ends].
[Figure caption and citation for the preceding image starts]: Retinite pigmentosaDo acervo do Oregon Retinal Degeneration Center [Citation ends].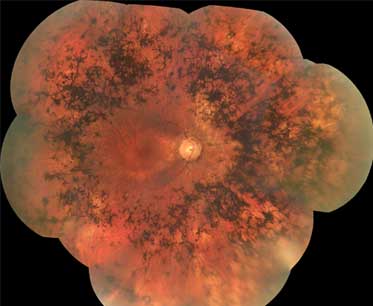
Drusas da cabeça do nervo óptico: drusas do disco óptico são anomalias congênitas e do desenvolvimento da cabeça do nervo óptico. Elas são formadas por degeneração cálcica em alguns dos axônios do nervo óptico. Frequentemente, elas são um achado incidental, mas são mais comuns em pacientes com RP. Com o tempo, elas podem crescer e invadir a camada de fibras nervosas da retina, causando defeitos no campo de visão. Uma razão escavação/disco pequena pode indicar drusas ópticas encobertas.
Edema macular cistoide (EMC): trata-se de uma complicação comum da RP e se parece com um pequeno cisto na fóvea quando o fundo é observado em alta magnificação usando-se uma lâmpada de fenda.
Células vítreas leves: elas podem ser observadas por um especialista em um exame dos olhos com lâmpada de fenda. Uma inflamação mais significativa deve levar à suspeita de outras doenças que podem mimetizar a RP.
Retinopatia tipo Coats: um sinal raro observado em certos pacientes com RP. Esta retinopatia caracteriza-se por telangiectasia vascular anormal em áreas periféricas da retina, que podem extravasar e causar exsudação na retina.
Ceratocone e glaucoma
Mais comum em pacientes com RP que na população geral.
O ceratocone é uma distorção visual decorrente de alterações estruturais na córnea, que a deixa com uma aparência mais cônica. Isso pode afetar a acuidade visual, e, frequentemente, a visão noturna é particularmente prejudicada.
O glaucoma é o resultado da elevação aguda ou crônica da pressão intraocular, causando dano ao nervo óptico. Ele pode ser detectado através da medição da pressão intraocular.
Investigações
O número de exames varia entre os centros, mas todos os pacientes com suspeita de RP devem passar por um teste do campo visual e fazer um eletrorretinograma (ERG).
Exame dos campos visuais: pode ser realizado de diferentes formas, e os testes dependem de se o estímulo é estático ou móvel (cinético). A perimetria cinética de Goldmann é um dos poucos testes que avalia todo o campo visual de um paciente. Ela fornece informações essenciais sobre a capacidade funcional, como a habilidade de dirigir com segurança. No entanto, o teste de campo visual com Octopus 900 hoje é capaz de realizar uma perimetria total, tanto cinética quanto estática. Defeitos do campo visual centro-periféricos são uma das características patognomônicas da RP. Os defeitos podem começar como ilhas na porção centro-periférica, se expandindo até que formem crescentes, resultando, por fim, em um escotoma anular completo. Com o tempo, esses escotomas anulares podem aumentar até deixar os pacientes com um campo de visão reduzido em túnel.
EGR de campo cheio: esse exame mede a resposta elétrica das células na retina, incluindo os fotorreceptores. Ele envolve a colocação de eletrodos na córnea ou na pele ao redor do olho para medir a amplitude das respostas a estímulos padrão, como um flash de luz. ERGs anormais são uma característica essencial da RP. Uma diminuição na amplitude da onda-b do cone e um aumento na latência podem ser observados nos ERGs adaptados à luz e também nos adaptados ao escuro.[31]
Exames adicionais que podem ser realizados para confirmar o diagnóstico incluem o teste de limiar elevado de adaptação ao escuro e a tomografia de coerência óptica (TCO). Essas técnicas não são realizadas rotineiramente em todos os centros.
O teste de limiar elevado de adaptação envolve a colocação do paciente em um quarto completamente escuro para determinar a mínima condição de luz que pode ser percebida. Ele replica relatos sintomáticos de dificuldade de adaptação a ambientes escurecidos.
A TCO da retina é um sistema de imagem não invasivo que usa a detecção da reflexão óptica da luz para construir uma imagem tridimensional. O exame não envolve contato e nem exposição à radiação. Ela pode revelar atrofia retiniana e é o método preferido para a determinação da presença de EMC. A TCO deve ser considerada para qualquer paciente com acuidade visual central diminuída sugestiva de EMC.
A imagiologia óptica adaptativa é uma nova tecnologia que permite obter imagens de alta resolução do mosaico dos fotorreceptores por compensação das aberrações lenticulares e corneanas durante o exame. Sistemas personalizados são capazes de revelar cones e bastonetes individuais em alguns pacientes.
O teste genético para RP envolve a coleta de sangue ou a obtenção de um swab bucal do paciente e o envio para um laboratório que testa mutações específicas para essa doença. Testes genéticos estão disponíveis apenas para uma parte dos genes relacionados à RP, e mesmo o teste para genes conhecidos nem sempre é completamente sensível. Eles são mais eficazes quando há suspeita de um gene em particular. Isso é realizado apenas em certas pessoas após a consulta com um geneticista clínico quando a probabilidade de identificar o gene responsável é alta. O resultado poderia confirmar o diagnóstico. Novas tecnologias que usam o sequenciamento completo do exoma estão disponíveis e permitem testar vários genes ao mesmo tempo. Além disso, essas tecnologias oferecem a esperança de encontrar novos genes para RP.
A autofluorescência de campo amplo do fundo (FAF) do olho é uma modalidade de exame de imagem que identifica as áreas do fundo do olho com distribuição irregular da lipofuscina e outros fluoróforos na monocamada de células do epitélio pigmentar da retina. Os padrões da FAF no polo posterior e na retina periférica estão associados a degenerações hereditárias específicas da retina e podem ser úteis no monitoramento da evolução da doença e da resposta a novas terapias. As diretrizes da American Academy of Ophthalmology fornecem recomendações para a avaliação clínica de pacientes com degenerações retinianas hereditárias.[31]
O uso deste conteúdo está sujeito ao nosso aviso legal