Etiologia
Genética
É amplamente aceito que uma mutação nos dois alelos do gene RB1 seja um pré-requisito para a presença da doença. É provável que sejam necessárias outras mutações para que ocorra a progressão para o retinoblastoma clínico.[24]
Um estudo demonstrou que os perfis de expressão gênica podem ser dinâmicos, variando com o tempo e a região (regiões tumorais apicais e basais).[25] No entanto, apenas 10% dos pacientes têm uma história familiar previamente estabelecida da doença.[24][26]
A maioria dos casos ocorre como resultado de mutações espontâneas inicialmente na embriogênese ou primariamente (de novo) em um conjunto de células das linhas germinativas parentais.[26]
Exposição viral
Os tipos 16 e 18 do papilomavírus humano (HPV) têm sido relatados em amostras de retinoblastoma.[27][28] Mais pesquisas são necessárias para determinar se há uma associação significativa entre o HPV e o retinoblastoma.[29][30]
Idade paterna avançada
Fisiopatologia
Seja herdado na linha germinativa ou ocorrendo espontaneamente, o retinoblastoma se desenvolve a partir de uma mutação crítica no gene RB1, que é expresso principalmente em interneurônios horizontais localizados na camada nuclear interna da retina.[36] Essa mutação causa a perda de heterozigosidade e subsequente inativação do RB1 (isto é, a formação de um mutante nulo homozigoto não funcional).[24]
O gene RB1 desempenha um papel fundamental como supressor de tumores, regulando o ciclo celular ao inibir a transição da fase G1 para a fase S.[37][38] Sua perda remove um checkpoint fundamental na replicação celular, resultando em proliferação celular descontrolada. A presença de outras membras da família de proteínas RB, como a p107 e a p130, e proteínas não relacionadas, como proteínas associadas à senescência, pode compensar parcialmente a perda do RB1 e levar a um retardo na progressão do tumor em alguns casos.[39] Entretanto, esses mecanismos compensatórios muitas vezes são insuficientes para prevenir a tumorigênese.
A falta de um RB1 funcional cria uma cascata de efeitos a jusante, incluindo instabilidade genômica, segregação cromossômica aberrante e perturbações na expressão de proteínas críticas para o controle do ciclo celular, como os fatores de transcrição E2F.[37][39] Essa desestabilização promove um ambiente propício à oncogênese. À medida que as mutações se acumulam, as células do retinoblastoma adquirem características adicionais de neoplasia maligna, como resistência à apoptose, angiogênese e evasão da vigilância imunológica.
Com o tempo, essas alterações celulares culminam na manifestação clínica do retinoblastoma, o qual pode ser detectado por meio de várias técnicas diagnósticas, incluindo exame de fundo de olho, exames de imagem e testes genéticos.[40]
[Figure caption and citation for the preceding image starts]: Histopatologia do retinoblastoma. Esta imagem demonstra as características clássicas do retinoblastoma, incluindo células tumorais pequenas, arredondadas e densamente concentradas, com núcleos hipercromáticos e citoplasma escasso, dispostas em camadas. A ausência de rosetas de Flexner-Wintersteiner nesta amostra não impede o diagnóstico, pois sua presença não é obrigatória. Essas características histopatológicas são típicas desse tumor retiniano agressivo e fornecem informações críticas para o diagnóstico, o estadiamento e o prognósticoPR J. L. Kemeny, ISM/Science Photo Library; usado com permissão [Citation ends].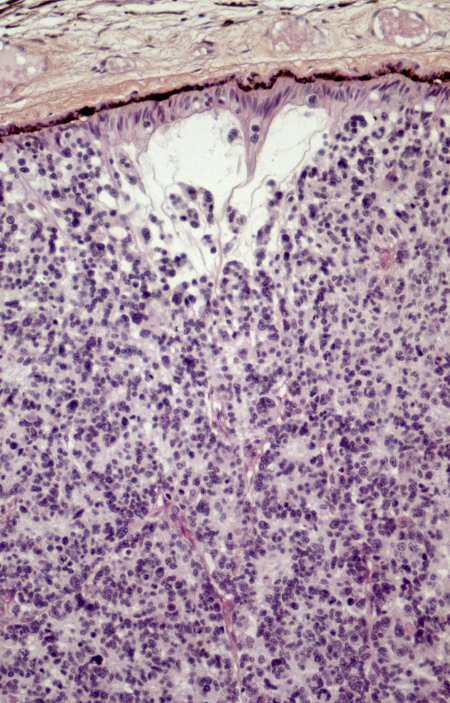 [Figure caption and citation for the preceding image starts]: Micrografia óptica de um corte da retina de um paciente com retinoblastoma, uma forma rara de câncer intraocular. O tumor apresenta uma arquitetura retiniana alterada e crescimento infiltrante de células atípicas. A imuno-histoquímica com anticorpos anti-rodopsina destaca áreas de diferenciação de fotorreceptores preservadas dentro do tecido retiniano. Essa coloração auxilia na identificação das camadas residuais da retina em meio à proliferação celular malignaPR J. L. Kemeny, ISM/Science Photo Library; usado com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Micrografia óptica de um corte da retina de um paciente com retinoblastoma, uma forma rara de câncer intraocular. O tumor apresenta uma arquitetura retiniana alterada e crescimento infiltrante de células atípicas. A imuno-histoquímica com anticorpos anti-rodopsina destaca áreas de diferenciação de fotorreceptores preservadas dentro do tecido retiniano. Essa coloração auxilia na identificação das camadas residuais da retina em meio à proliferação celular malignaPR J. L. Kemeny, ISM/Science Photo Library; usado com permissão [Citation ends].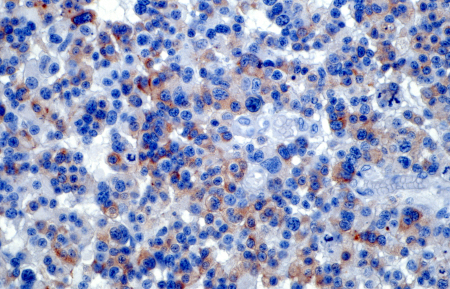
Classificação
Classificação internacional de retinoblastoma[6]
O objetivo desta classificação é refletir a probabilidade de sobrevivência ocular com base em técnicas de tratamento modernas (geralmente, quimioterapia associada a terapia focal).
Grupo A
Retinoblastoma de 3 mm ou menos de dimensão basal ou espessura, localizado, pelo menos, a 3 mm da fovéola e 1.5 mm do nervo óptico.
Grupo B
Retinoblastoma não incluído no Grupo A com um ou mais dos seguintes sinais:
Localização macular (≤3 mm de distância da fovéola)
Localização justapapilar (≤1.5 mm de distância do nervo óptico)
Fluido sub-retiniano adicional (≤5 mm da margem).
Grupo C
Tumor de retinoblastoma com um dos seguintes sinais:
Acometimento sub-retiniano focal
Acometimento vítreo focal
Acometimento vítreo e sub-retiniano focal.
Grupo D
Tumor de retinoblastoma com um dos seguintes sinais:
Acometimento sub-retiniano difuso
Acometimento vítreo difuso
Acometimento vítreo e sub-retiniano difuso.
Grupo E
Olhos com risco muito alto, apresentando um ou mais dos seguintes sinais:
Glaucoma neovascular
Hemorragia intraocular densa
Celulite orbitária asséptica
Tumor anterior à face vítrea
Tumor que toca o cristalino
Retinoblastoma infiltrante difuso
Atrofia degenerativa do globo ocular (com hipotonia) (também conhecida como doença terminal com acometimento ocular, na qual o globo ocular está não funcional, desorganizado, apresenta cicatrizes e frequentemente calcificação distrófica).
Classificação de Reese-Ellsworth[7]
Reflete a probabilidade de sobrevivência ocular após radiação por feixe externo pela parede lateral. A classificação de Reese-Ellsworth foi amplamente substituída pela International Intraocular Retinoblastoma Classification (após a introdução da quimioterapia intravenosa para o retinoblastoma intraocular).
Grupo I
a. Tumor solitário, <4 diâmetros de disco de tamanho, localizado na linha do equador do olho (linha imaginária no plano coronal que marca a divisão entre as metades anterior e posterior do olho) ou posterior a ela.
b. Vários tumores, nenhum >4 diâmetros de disco de tamanho, todos na linha do equador ou atrás dela.
Grupo II
b. Tumor solitário, 4 a 10 diâmetros de disco de tamanho, localizado na linha do equador ou atrás dela.
b. Vários tumores, 4 a 10 diâmetros de disco de tamanho, localizados atrás da linha do equador.
Grupo III
a. Qualquer lesão anterior à linha do equador.
b. Tumores solitários >10 diâmetros de disco, localizados atrás da linha do equador.
Grupo IV
a. Vários tumores, alguns >10 diâmetros de disco de tamanho.
b. Qualquer lesão que se estende anteriormente à ora serrata (a junção denteada entre a retina e o corpo ciliar).
Grupo V
a. Tumores maciços que envolvem a metade da retina.
b. Acometimento vítreo.
O uso deste conteúdo está sujeito ao nosso aviso legal