El diagnóstico de SMD requiere una historia clínica y una exploración física detalladas, y una evaluación patológica de la sangre periférica y la médula ósea.
Los SMD son una enfermedad heterogénea con presentaciones variables. Los pacientes suelen estar asintomáticos en el momento de la presentación y se sospecha SMD después de un análisis de sangre de rutina que muestra citopenia (más comúnmente anemia).[47]Garcia-Manero G. Myelodysplastic syndromes: 2023 update on diagnosis, risk-stratification, and management. Am J Hematol. 2023 Aug;98(8):1307-25.
https://www.doi.org/10.1002/ajh.26984
http://www.ncbi.nlm.nih.gov/pubmed/37288607?tool=bestpractice.com
Algunos pacientes presentan síntomas relacionados con la citopenia (p. ej., fatiga, infecciones, hematomas).
Anamnesis y exploración física
La mediana de edad en el momento del diagnóstico es de 70 a 75 años, pero la enfermedad puede ocurrir a cualquier edad y debe considerarse en pacientes más jóvenes que hayan estado expuestos previamente a quimioterapia o radioterapia, o que tengan un trastorno congénito (p. ej., síndrome de Fanconi, síndrome de Bloom, Síndrome de Down).[9]Sekeres MA, Taylor J. Diagnosis and treatment of myelodysplastic syndromes: a review. JAMA. 2022 Sep 6;328(9):872-80.
http://www.ncbi.nlm.nih.gov/pubmed/36066514?tool=bestpractice.com
[10]Roman E, Smith A, Appleton S, et al. Myeloid malignancies in the real-world: occurrence, progression and survival in the UK's population-based Haematological Malignancy Research Network 2004-15. Cancer Epidemiol. 2016 Jun;42:186-98.
https://www.sciencedirect.com/science/article/pii/S1877782116300364
http://www.ncbi.nlm.nih.gov/pubmed/27090942?tool=bestpractice.com
[14]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: myelodysplastic syndromes [internet publication].
https://www.nccn.org/guidelines/category_1
[15]Fenaux P, Haase D, Santini V, et al; ESMO Guidelines Committee. Myelodysplastic syndromes: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2021 Feb;32(2):142-56.
https://www.annalsofoncology.org/article/S0923-7534(20)43129-1/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/33221366?tool=bestpractice.com
[17]Sperling AS, Gibson CJ, Ebert BL. The genetics of myelodysplastic syndrome: from clonal haematopoiesis to secondary leukaemia. Nat Rev Cancer. 2017 Jan;17(1):5-19.
http://www.ncbi.nlm.nih.gov/pubmed/27834397?tool=bestpractice.com
[25]Oetjen KA, Levoska MA, Tamura D, et al. Predisposition to hematologic malignancies in patients with xeroderma pigmentosum. Haematologica. 2020 Apr;105(4):e144-6.
https://www.doi.org/10.3324/haematol.2019.223370
http://www.ncbi.nlm.nih.gov/pubmed/31439674?tool=bestpractice.com
[26]Aktas D, Koc A, Boduroglu K, et al. Myelodysplastic syndrome associated with monosomy 7 in a child with Bloom syndrome. Cancer Genet Cytogenet. 2000 Jan;116(1):44-6.
http://www.ncbi.nlm.nih.gov/pubmed/10616531?tool=bestpractice.com
[35]Sill H, Olipitz W, Zebisch A, et al. Therapy-related myeloid neoplasms: pathobiology and clinical characteristics. Br J Pharmacol. 2011 Feb;162(4):792-805.
https://bpspubs.onlinelibrary.wiley.com/doi/10.1111/j.1476-5381.2010.01100.x
http://www.ncbi.nlm.nih.gov/pubmed/21039422?tool=bestpractice.com
[36]Kaplan H, Malmgren J, De Roos AJ. Risk of myelodysplastic syndrome and acute myeloid leukemia post radiation treatment for breast cancer: a population-based study. Breast Cancer Res Treat. 2013 Feb;137(3):863-7.
http://www.ncbi.nlm.nih.gov/pubmed/23274844?tool=bestpractice.com
[48]Sallman DA, Padron E. Myelodysplasia in younger adults: outlier or unique molecular entity? Haematologica. 2017 Jun;102(6):967-8.
https://www.doi.org/10.3324/haematol.2017.165993
http://www.ncbi.nlm.nih.gov/pubmed/28566339?tool=bestpractice.com
[49]Hirsch CM, Przychodzen BP, Radivoyevitch T, et al. Molecular features of early onset adult myelodysplastic syndrome. Haematologica. 2017 Jun;102(6):1028-34.
https://www.doi.org/10.3324/haematol.2016.159772
http://www.ncbi.nlm.nih.gov/pubmed/28255022?tool=bestpractice.com
Los antecedentes deben incluir una evaluación detallada de la exposición previa a quimioterapia y/o radioterapia; infecciones previas o episodios de sangrado; presencia de comorbilidades; antecedentes familiares de trastornos hematológicos; estado nutricional (deficiencias de nutrientes); consumo de alcohol; y exposición a sustancias químicas tóxicas.[11]Killick SB, Wiseman DH, Quek L, et al. British Society for Haematology guidelines for the diagnosis and evaluation of prognosis of adult myelodysplastic syndromes. Br J Haematol. 2021 Jul;194(2):282-93.
https://onlinelibrary.wiley.com/doi/10.1111/bjh.17621
http://www.ncbi.nlm.nih.gov/pubmed/34137023?tool=bestpractice.com
[14]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: myelodysplastic syndromes [internet publication].
https://www.nccn.org/guidelines/category_1
Se requiere una exploración física detallada que pueda identificar signos y síntomas relacionados con las citopenias, como palidez, fatiga, intolerancia al ejercicio, infecciones (generalmente bacterianas), hematomas y sangrado (petequias, púrpura).
Se informan trastornos autoinmunitarios (p. ej., vasculitis, enfermedad del tejido conjuntivo, artritis inflamatoria) en aproximadamente el 25% de los pacientes con SMD.[6]Wolach O, Stone R. Autoimmunity and inflammation in myelodysplastic syndromes. Acta Haematol. 2016;136(2):108-17.
https://www.doi.org/10.1159/000446062
http://www.ncbi.nlm.nih.gov/pubmed/27337745?tool=bestpractice.com
[7]Komrokji RS, Kulasekararaj A, Al Ali NH, et al. Autoimmune diseases and myelodysplastic syndromes. Am J Hematol. 2016 May;91(5):E280-3.
https://www.doi.org/10.1002/ajh.24333
http://www.ncbi.nlm.nih.gov/pubmed/26875020?tool=bestpractice.com
[8]Enright H, Jacobs HS, Vercellotti G, et al. Paraneoplastic autoimmune phenomena in patients with myelodysplastic syndromes: response to immunosuppressive therapy. Br J Haematol. 1995 Oct;91(2):403-8.
http://www.ncbi.nlm.nih.gov/pubmed/8547082?tool=bestpractice.com
En los SMD rara vez se presentan esplenomegalia, hepatomegalia y linfadenopatía. Pueden aparecer en la leucemia mielomonocítica crónica (LMMC), una neoplasia mieloide con características patológicas y moleculares que se solapan con los SMD.[1]Khoury JD, Solary E, Abla O, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. 2022 Jul;36(7):1703-19.
https://www.doi.org/10.1038/s41375-022-01613-1
http://www.ncbi.nlm.nih.gov/pubmed/35732831?tool=bestpractice.com
Pruebas iniciales
Las pruebas iniciales deben ser un hemograma completo (HC) con diferencial y un frotis de sangre periférica. El HC mostrará una o más citopenias (más comúnmente anemia) que son sostenidas (p. ej., >4 meses).[11]Killick SB, Wiseman DH, Quek L, et al. British Society for Haematology guidelines for the diagnosis and evaluation of prognosis of adult myelodysplastic syndromes. Br J Haematol. 2021 Jul;194(2):282-93.
https://onlinelibrary.wiley.com/doi/10.1111/bjh.17621
http://www.ncbi.nlm.nih.gov/pubmed/34137023?tool=bestpractice.com
[14]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: myelodysplastic syndromes [internet publication].
https://www.nccn.org/guidelines/category_1
[15]Fenaux P, Haase D, Santini V, et al; ESMO Guidelines Committee. Myelodysplastic syndromes: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2021 Feb;32(2):142-56.
https://www.annalsofoncology.org/article/S0923-7534(20)43129-1/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/33221366?tool=bestpractice.com
El frotis de sangre periférica mostrará citopenias y displasia (p. ej., granulocitos hipogranulares e hipolobulados [anomalía pseudo-Pelger-Huet]).[15]Fenaux P, Haase D, Santini V, et al; ESMO Guidelines Committee. Myelodysplastic syndromes: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2021 Feb;32(2):142-56.
https://www.annalsofoncology.org/article/S0923-7534(20)43129-1/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/33221366?tool=bestpractice.com
Los análisis clínicos adicionales incluyen recuento de reticulocitos, folato de eritrocitos, vitamina B12 sérica y análisis de hierro (hierro sérico, capacidad total de fijación del hierro, ferritina).[11]Killick SB, Wiseman DH, Quek L, et al. British Society for Haematology guidelines for the diagnosis and evaluation of prognosis of adult myelodysplastic syndromes. Br J Haematol. 2021 Jul;194(2):282-93.
https://onlinelibrary.wiley.com/doi/10.1111/bjh.17621
http://www.ncbi.nlm.nih.gov/pubmed/34137023?tool=bestpractice.com
[14]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: myelodysplastic syndromes [internet publication].
https://www.nccn.org/guidelines/category_1
[15]Fenaux P, Haase D, Santini V, et al; ESMO Guidelines Committee. Myelodysplastic syndromes: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2021 Feb;32(2):142-56.
https://www.annalsofoncology.org/article/S0923-7534(20)43129-1/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/33221366?tool=bestpractice.com
Deben realizarse para descartar otras causas de citopenias. El recuento de reticulocitos suele ser bajo en los SMD.[50]Juneja SK, Imbert M, Jouault H, et al. Haematological features of primary myelodysplastic syndromes (PMDS) at initial presentation: a study of 118 cases. J Clin Pathol. 1983 Oct;36(10):1129-35.
https://jcp.bmj.com/content/jclinpath/36/10/1129.full.pdf
http://www.ncbi.nlm.nih.gov/pubmed/6619310?tool=bestpractice.com
Se pueden realizar pruebas de infección viral (p. ej., VIH; hepatitis B, C y E; citomegalovirus; parvovirus) si existen factores de riesgo de exposición previa.[11]Killick SB, Wiseman DH, Quek L, et al. British Society for Haematology guidelines for the diagnosis and evaluation of prognosis of adult myelodysplastic syndromes. Br J Haematol. 2021 Jul;194(2):282-93.
https://onlinelibrary.wiley.com/doi/10.1111/bjh.17621
http://www.ncbi.nlm.nih.gov/pubmed/34137023?tool=bestpractice.com
[14]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: myelodysplastic syndromes [internet publication].
https://www.nccn.org/guidelines/category_1
[15]Fenaux P, Haase D, Santini V, et al; ESMO Guidelines Committee. Myelodysplastic syndromes: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2021 Feb;32(2):142-56.
https://www.annalsofoncology.org/article/S0923-7534(20)43129-1/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/33221366?tool=bestpractice.com
La infección por VIH puede causar cambios displásicos en la médula ósea similares a los observados en los SMD.[51]Katsarou O, Terpos E, Patsouris E, et al. Myelodysplastic features in patients with long-term HIV infection and haemophilia. Haemophilia. 2001 Jan;7(1):47-52.
https://onlinelibrary.wiley.com/doi/abs/10.1111/j.1365-2516.2001.00445.x?sid=nlm%3Apubmed
http://www.ncbi.nlm.nih.gov/pubmed/11136381?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: Película de sangre que muestra neutrófilos normales (derecha) y neutrófilos displásicos con citoplasma agranular y núcleo hipolobadoImagen usada con permiso de BMJ 1997;314:883 [Citation ends].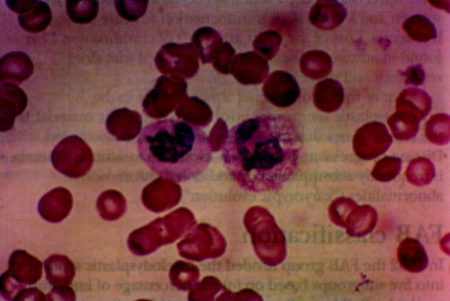
Evaluación de la médula ósea
Se requieren aspiración de médula ósea (con tinción de hierro) y biopsia con aguja gruesa para análisis morfológicos, citogenéticos, mutacionales y de citometría de flujo.[14]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: myelodysplastic syndromes [internet publication].
https://www.nccn.org/guidelines/category_1
[47]Garcia-Manero G. Myelodysplastic syndromes: 2023 update on diagnosis, risk-stratification, and management. Am J Hematol. 2023 Aug;98(8):1307-25.
https://www.doi.org/10.1002/ajh.26984
http://www.ncbi.nlm.nih.gov/pubmed/37288607?tool=bestpractice.com
Estas pruebas diagnósticas confirman el diagnóstico de SMD y guían la estratificación y el manejo del riesgo.[14]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: myelodysplastic syndromes [internet publication].
https://www.nccn.org/guidelines/category_1
[47]Garcia-Manero G. Myelodysplastic syndromes: 2023 update on diagnosis, risk-stratification, and management. Am J Hematol. 2023 Aug;98(8):1307-25.
https://www.doi.org/10.1002/ajh.26984
http://www.ncbi.nlm.nih.gov/pubmed/37288607?tool=bestpractice.com
[52]van de Loosdrecht AA, Kern W, Porwit A, et al. Clinical application of flow cytometry in patients with unexplained cytopenia and suspected myelodysplastic syndrome: a report of the European LeukemiaNet International MDS-Flow Cytometry Working Group. Cytometry B Clin Cytom. 2023 Jan;104(1):77-86.
https://onlinelibrary.wiley.com/doi/10.1002/cyto.b.22044
http://www.ncbi.nlm.nih.gov/pubmed/34897979?tool=bestpractice.com
Se puede realizar un diagnóstico de SMD en un paciente con citopenia persistente en presencia de uno de los tres criterios siguientes: displasia significativa de la médula ósea (≥10% en uno o más de los tres linajes principales de la médula ósea); blastos en sangre periférica y/o médula ósea (<20%); o una anomalía citogenética clonal o una mutación somática.[1]Khoury JD, Solary E, Abla O, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. 2022 Jul;36(7):1703-19.
https://www.doi.org/10.1038/s41375-022-01613-1
http://www.ncbi.nlm.nih.gov/pubmed/35732831?tool=bestpractice.com
[2]Arber DA, Orazi A, Hasserjian RP, et al. International Consensus Classification of myeloid neoplasms and acute leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022 Sep 15;140(11):1200-28.
https://www.doi.org/10.1182/blood.2022015850
http://www.ncbi.nlm.nih.gov/pubmed/35767897?tool=bestpractice.com
[11]Killick SB, Wiseman DH, Quek L, et al. British Society for Haematology guidelines for the diagnosis and evaluation of prognosis of adult myelodysplastic syndromes. Br J Haematol. 2021 Jul;194(2):282-93.
https://onlinelibrary.wiley.com/doi/10.1111/bjh.17621
http://www.ncbi.nlm.nih.gov/pubmed/34137023?tool=bestpractice.com
Las características biológicas son más importantes que un valor límite estricto de blastos.[14]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: myelodysplastic syndromes [internet publication].
https://www.nccn.org/guidelines/category_1
Los pacientes con blastos ≥20% deben ser evaluados para detectar leucemia mieloide aguda. (Ver Leucemia mieloide aguda).
[Figure caption and citation for the preceding image starts]: Megacariocito mononuclear grande en la médula ósea de un paciente con SMD-del(5q)Imagen usada con permiso de BMJ 1997;314:883 [Citation ends].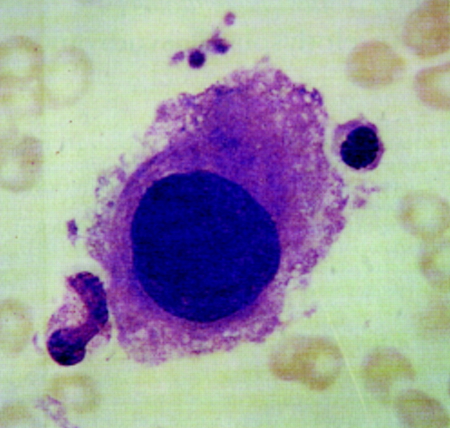
Pruebas genéticas
Pruebas genéticas para detectar alteraciones citogenéticas asociadas a SMD (p. ej., -5, del(5q), -7, del(7q), del(11q), del(12p), -17, del(17p), del(20q)) y las mutaciones somáticas (p. ej., DNMT3A, TET2, ASXL1, RUNX1, SRSF2, TP53, SF3B1) informan el diagnóstico y la estratificación del riesgo pronóstico.[11]Killick SB, Wiseman DH, Quek L, et al. British Society for Haematology guidelines for the diagnosis and evaluation of prognosis of adult myelodysplastic syndromes. Br J Haematol. 2021 Jul;194(2):282-93.
https://onlinelibrary.wiley.com/doi/10.1111/bjh.17621
http://www.ncbi.nlm.nih.gov/pubmed/34137023?tool=bestpractice.com
[14]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: myelodysplastic syndromes [internet publication].
https://www.nccn.org/guidelines/category_1
Las mutaciones en el gen de la isocitrato deshidrogenasa 1 (IDH1) pueden orientar las elecciones de tratamiento.[11]Killick SB, Wiseman DH, Quek L, et al. British Society for Haematology guidelines for the diagnosis and evaluation of prognosis of adult myelodysplastic syndromes. Br J Haematol. 2021 Jul;194(2):282-93.
https://onlinelibrary.wiley.com/doi/10.1111/bjh.17621
http://www.ncbi.nlm.nih.gov/pubmed/34137023?tool=bestpractice.com
[14]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: myelodysplastic syndromes [internet publication].
https://www.nccn.org/guidelines/category_1
La presencia de determinadas alteraciones citogenéticas o mutaciones somáticas (p. ej., -7/del(7q), del(5q) y SF3B1) puede establecer un diagnóstico sin displasia.[1]Khoury JD, Solary E, Abla O, et al. The 5th edition of the World Health Organization classification of haematolymphoid tumours: myeloid and histiocytic/dendritic neoplasms. Leukemia. 2022 Jul;36(7):1703-19.
https://www.doi.org/10.1038/s41375-022-01613-1
http://www.ncbi.nlm.nih.gov/pubmed/35732831?tool=bestpractice.com
[2]Arber DA, Orazi A, Hasserjian RP, et al. International Consensus Classification of myeloid neoplasms and acute leukemias: integrating morphologic, clinical, and genomic data. Blood. 2022 Sep 15;140(11):1200-28.
https://www.doi.org/10.1182/blood.2022015850
http://www.ncbi.nlm.nih.gov/pubmed/35767897?tool=bestpractice.com
Se pueden realizar pruebas genéticas en sangre periférica si no es posible realizar pruebas de médula ósea.
Los pacientes con displasia significativa que no tienen una anomalía citogenética clonal o una mutación somática deben someterse a una evaluación adicional para descartar una causa no maligna de displasia.
Se deben realizar pruebas genéticas adicionales en pacientes con sospecha de síndrome de predisposición hereditaria (por ejemplo, pacientes de <50 años o con características clínicamente sugestivas, o pruebas somáticas sugestivas de mutación de estirpe germinal).[14]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: myelodysplastic syndromes [internet publication].
https://www.nccn.org/guidelines/category_1
Pruebas subsiguientes
Una vez que se haya establecido el diagnóstico, en algunas situaciones pueden resultar útiles las siguientes pruebas adicionales.
Niveles de eritropoyetina sérica: se pueden medir para guiar el tratamiento con agentes estimulantes de la eritropoyesis.[14]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: myelodysplastic syndromes [internet publication].
https://www.nccn.org/guidelines/category_1
[15]Fenaux P, Haase D, Santini V, et al; ESMO Guidelines Committee. Myelodysplastic syndromes: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2021 Feb;32(2):142-56.
https://www.annalsofoncology.org/article/S0923-7534(20)43129-1/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/33221366?tool=bestpractice.com
[53]de Witte T, Bowen D, Robin M, et al. Allogeneic hematopoietic stem cell transplantation for MDS and CMML: recommendations from an international expert panel. Blood. 2017 Mar 30;129(13):1753-62.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5524528
http://www.ncbi.nlm.nih.gov/pubmed/28096091?tool=bestpractice.com
La eritropoyetina sérica suele estar elevada en los SMD, excepto en caso de insuficiencia renal concurrente, en cuyo caso es baja.
Lactato deshidrogenasa: tiene valor pronóstico y puede medirse para informar la estratificación y el manejo del riesgo.[14]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: myelodysplastic syndromes [internet publication].
https://www.nccn.org/guidelines/category_1
[15]Fenaux P, Haase D, Santini V, et al; ESMO Guidelines Committee. Myelodysplastic syndromes: ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2021 Feb;32(2):142-56.
https://www.annalsofoncology.org/article/S0923-7534(20)43129-1/fulltext
http://www.ncbi.nlm.nih.gov/pubmed/33221366?tool=bestpractice.com
La lactato deshidrogenasa elevada se asocia con desenlaces más desfavorables.[54]Wimazal F, Sperr WR, Kundi M, et al. Prognostic value of lactate dehydrogenase activity in myelodysplastic syndromes. Leuk Res. 2001 Apr;25(4):287-94.
https://www.doi.org/10.1016/s0145-2126(00)00140-5
http://www.ncbi.nlm.nih.gov/pubmed/11248325?tool=bestpractice.com
[55]Germing U, Hildebrandt B, Pfeilstöcker M, et al. Refinement of the international prognostic scoring system (IPSS) by including LDH as an additional prognostic variable to improve risk assessment in patients with primary myelodysplastic syndromes (MDS). Leukemia. 2005 Dec;19(12):2223-31.
http://www.ncbi.nlm.nih.gov/pubmed/16193087?tool=bestpractice.com
La tipificación de HLA: es útil si el paciente es candidato para un trasplante de células madre hematopoyéticas o si se necesitan o anticipan transfusiones de plaquetas frecuentes.[14]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: myelodysplastic syndromes [internet publication].
https://www.nccn.org/guidelines/category_1
[53]de Witte T, Bowen D, Robin M, et al. Allogeneic hematopoietic stem cell transplantation for MDS and CMML: recommendations from an international expert panel. Blood. 2017 Mar 30;129(13):1753-62.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5524528
http://www.ncbi.nlm.nih.gov/pubmed/28096091?tool=bestpractice.com
Citometría de flujo: puede contribuir al diagnóstico (identificando características displásicas y blastos) y pronóstico. Puede usarse (junto con la prueba de mutación STAT3) para la evaluación de un clon de hemoglobinuria paroxística nocturna concurrente y una posible leucemia de linfocitos grandes granulares.[3]Steensma DP, Bennett JM. The myelodysplastic syndromes: diagnosis and treatment. Mayo Clin Proc. 2006 Jan;81(1):104-30.
http://www.ncbi.nlm.nih.gov/pubmed/16438486?tool=bestpractice.com
[14]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: myelodysplastic syndromes [internet publication].
https://www.nccn.org/guidelines/category_1