Etiologia
A síndrome de DiGeorge (síndrome de deleção 22q11.2 ou 22q11.2DS) resulta de uma deleção de 1 cópia na região cromossômica 22q11.2.[18] A deleção resulta em hemizigosidade para pelo menos 30 genes, incluindo TBX1, um fator de transcrição importante para o desenvolvimento dos arcos faríngeos.[2][19] Essa deleção específica é muito mais comum do que outras microdeleções devido à estrutura genômica dessa região do cromossomo 22.[20] Duas áreas de repetições com baixo número de cópias ocorrem nos pontos de quebra da deleção; a deleção em si surge quando essas duas áreas se recombinam anormalmente durante a meiose.[21] A estrutura semelhante das repetições com baixo número de cópias resulta em pareamento incorreto e na deleção da porção do cromossomo entre as duas repetições. A deleção típica é de 3 megabases, embora 10% a 15% dos pacientes possam ter uma deleção menor de cerca de 1.5 megabase.[22] Essa deleção menor resulta da recombinação anormal com uma terceira repetição com baixo número de cópias. Embora essas deleções sejam a causa primária da síndrome, 2% dos pacientes apresentam pequenas deleções 22q11.2 atípicas, e alguns pacientes não apresentam deleção.[23][24][25][26][27][28] Constatou-se que alguns desses pacientes apresentam mutações pontuais em TBX1, enquanto outros apresentam deleções em 10p ou mutações na proteína de ligação ao cromodomínio-helicase DNA.[24][25][26] Também existem fenocópias da síndrome de DiGeorge, como as que podem ser encontradas em lactentes de mães diabéticas.[29] A variação genética em TBX1, o principal gene responsável pela síndrome, não provoca a variabilidade no fenótipo observada na síndrome.[30] Não existem fatores de risco modificáveis conhecidos para a deleção. A maioria dos casos de síndrome de DiGeorge é esporádica, mas as mutações conhecidas são hereditárias autossômicas dominantes.[14]
Fisiopatologia
A síndrome de DiGeorge é um conjunto de características morfológicas e neurológicas específicas que resultam da deleção de 1 cópia de 22q11.2.[18] A deleção provoca uma redução em TBX1, um fator de transcrição importante para o desenvolvimento dos arcos faríngeos.[2]
TBX1 interage com várias outras moléculas de sinalização, incluindo fatores de crescimento de fibroblastos e fatores de crescimento endotelial vascular, para promover o desenvolvimento do arco faríngeo.[31] Como TBX1 não é suficiente, os arcos e bolsas faríngeos são mal formados, e as estruturas que eles produzem são hipoplásicas.[32] Haploinsuficiência de TBX1 em camundongos é suficiente para afetar a artéria do quarto arco faríngeo, replicando uma característica importante da síndrome de DiGeorge.[32] Camundongos homozigotos para deleções de TBX1 mostram ausência total do timo e das glândulas paratireoides, e camundongos hemizigotos para a deleção sintênica apresentam timo anormal.[19][33] O TBX1 também é encontrado no campo cardíaco secundário e no cérebro, onde pode contribuir para o desenvolvimento do fenótipo neurológico.[34] Investigação em camundongos indica que, quando há deficiência de TBX1, todos os embriões desenvolvem inicialmente características do distúrbio, mas alguns parecem ser capazes de corrigir o defeito durante estágios posteriores do desenvolvimento.[33] Isso indica que outros genes podem estar envolvidos na modificação do distúrbio, e também pode sugerir que o desenvolvimento de pelo menos algumas características da doença seja evitável.
As investigações com o modelo de camundongo de deleção 22q11.2 sugeriram que as alterações no processamento de microRNA causadas pela deficiência de DGCR8, um gene encontrado na região de deleção, podem contribuir para as anormalidades neurológicas e as imagens cerebrais anormais observadas na síndrome de DiGeorge.[35][36]
Polimorfismos identificados em alguns genes parecem aumentar o risco de complicações específicas.[37][38][39]
Polimorfismos no gene catecol-O-metiltransferase na região deletada podem aumentar o risco de esquizofrenia, embora os estudos sejam conflitantes. Polimorfismos do fator de crescimento endotelial vascular alteram a incidência de cardiopatia em um modelo murino do distúrbio. Mecanismos semelhantes podem estar presentes em humanos. Esses polimorfismos ainda não estão disponíveis para testes clínicos ou para prever desfechos para pacientes.
Autoimunidade também é uma característica da deleção 22q11.2 e está correlacionada com a gravidade da linfopenia de células T.[40][Figure caption and citation for the preceding image starts]: Perda de 1 cópia de 22q11.2, demonstrada por análise do número de cópias por microarrayDos acervos de Sean A. McGhee, MD e Maria Garcia Lloret, MD [Citation ends].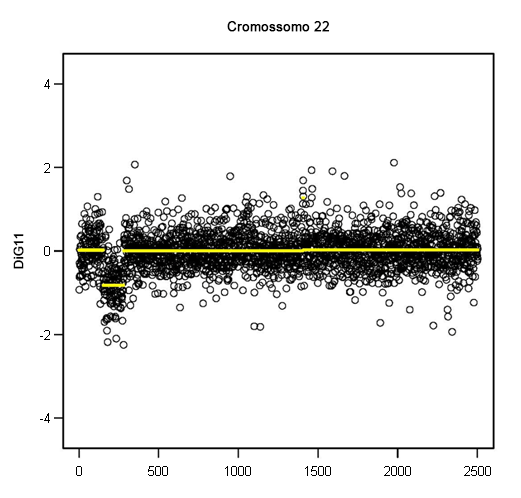
Classificação
Síndrome de DiGeorge completa e parcial (critérios de diagnóstico ESID/PAGID)[4]
Tradicionalmente, a síndrome de DiGeorge é dividida em síndrome de DiGeorge completa e parcial, embora uma avaliação mais detalhada da variabilidade da síndrome tenha diminuído a relevância dessa distinção.
O termo síndrome de DiGeorge completa era usado quando os pacientes apresentavam o espectro completo das manifestações típicas, incluindo imunodeficiência grave.
O termo síndrome de DiGeorge parcial era usado quando os pacientes apresentavam apenas algumas manifestações do distúrbio, principalmente aquelas sem imunodeficiência evidente. A síndrome de DiGeorge parcial é muito mais comum que a completa.
A medição da quantidade de células T virgens é fundamental para diferenciar síndrome de DiGeorge parcial e completa.[5]
Síndrome de DiGeorge típica e atípica[6]
Observações identificaram um pequeno subgrupo de pacientes com síndrome de DiGeorge que têm imunodeficiência de células T, mas desenvolvem células T oligoclonais anormais, o que resulta em uma erupção cutânea tipo doença do enxerto contra o hospedeiro e requer imunossupressão para evitar complicações de linfoproliferação.[6]
O termo síndrome de DiGeorge típica é usado quando os pacientes apresentam graus variados de imunodeficiência, mas nenhuma população de células T oligoclonais.
O termo síndrome de DiGeorge atípica é usado quando os pacientes apresentam proliferação de populações de células T oligoclonais autorreativas.
Essas duas distinções (típica e atípica) se aplicam apenas a pacientes com síndrome de DiGeorge completa, a qual se refere especificamente a imunodeficiência de células T grave (atimia).
A síndrome de DiGeorge também pode ser associada a anormalidades significativas das células B em uma parcela maior de pacientes do que se pensava anteriormente.[7][8]
O uso deste conteúdo está sujeito ao nosso aviso legal