História e exame físico
Principais fatores diagnósticos
comuns
história familiar
Se um membro da família com CET for um parente de primeiro grau (ou seja, um pai), existe uma chance de 50% de o paciente ter o distúrbio. Esse risco de 50% permanece quando vários irmãos são afetados, assumindo que um dos pais tem a doença sem saber ou que há mosaicismo gonadal em um dos pais. Essa é uma doença pela qual os únicos órgãos afetados pela mutação do CET são as gônadas e seus gametas afetados, permitindo o potencial para a existência de múltiplos filhos afetados de um pai ou mãe não afetado. Estima-se que a prevalência seja <2% de todos os pais com um filho afetado.[2][3][18]
No entanto, a maioria (cerca de dois terços) das pessoas com CET tem ocorrência esporádica.[4] Uma história familiar de três gerações deve ser obtida para avaliar os membros adicionais da família em risco de CET.[1]
epilepsia
A epilepsia, o sintoma manifesto mais comum da doença, ocorre em até 60% a 90% dos pacientes com CET ao longo da vida.[21]
Todos os tipos de convulsão podem se desenvolver, com exceção da pura epilepsia tipo ausência.
Na maioria das vezes, a epilepsia se manifesta na infância e o sintoma manifesto pode ser espasmos infantis em um terço dos pacientes.[2][3][22] Infantile Spasms Action Network: what does IS look like? Opens in new window
rabdomioma cardíaco (pode ser único ou múltiplo)
Afeta aproximadamente 60% das crianças e 20% dos adultos com CET.[23] Entre os lactentes com múltiplas lesões, 80% ou mais são eventualmente diagnosticados com CET. Essas lesões podem ser identificadas na 22ª semana de gestação. A maioria dos pacientes tem uma média de 3 lesões, de 3 a 25 mm, mas raramente maiores. Elas são identificadas nos ventrículos mais frequentemente que nos átrios, e no septo mais frequentemente que nas paredes.
Os sintomas (arritmias, obstrução do fluxo de saída, tromboembolismo) são raros e tipicamente evidentes no período neonatal. Essas lesões geralmente regridem durante a primeira infância sem a necessidade de intervenção.[2][3][24][Figure caption and citation for the preceding image starts]: Rabdomioma cardíaco na ressonância nuclear magnética (RNM) sagital em T1Cortesia do Dr. Francis J. DiMario Jr [Citation ends].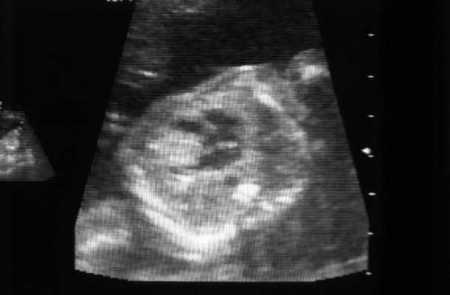
angiomiolipomas renais
Um estudo em grande escala revelou que os angiomiolipomas (AML) renais são relatados em quase 50% dos pacientes, e nos pacientes com AML renal presente, 88% apresentavam lesões múltiplas, 84% apresentavam lesões bilaterais, 33% apresentavam lesões maiores que 3 cm, e 21% apresentavam lesões crescentes.[26] Um quarto dos pacientes também apresentava cistos renais.[26]
Os AMLs aumentam em tamanho e número ao longo da vida, ao passo que os cistos podem regredir. Múltiplos cistos se assemelham à doença renal policística, sendo os cistos constituídos de células epiteliais eosinofílicas hipertróficas. Os AMLs são constituídos de uma mistura benigna de vasos sanguíneos, músculo liso e gordura. Raramente ocorre um AML maligno ou carcinoma de células renais.[26]
A maioria dos AMLs produz dor e hemorragia na vida adulta, ao passo que os cistos mais provavelmente causam hipertensão e insuficiência renal. Ocorre, como um todo, grave comprometimento renal em <5% dos pacientes.[2][3][24][Figure caption and citation for the preceding image starts]: Múltiplos angiomiolipomas renais na tomografia computadorizada (TC) axialCortesia do Dr. Francis J. DiMario Jr [Citation ends].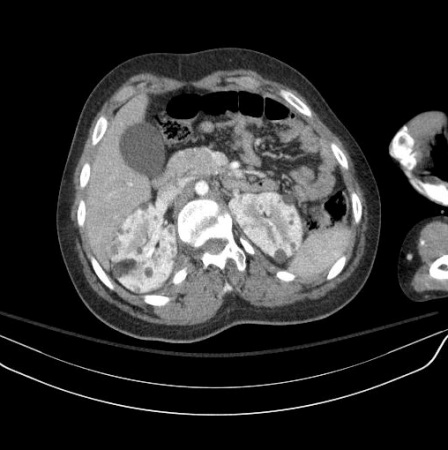 [Figure caption and citation for the preceding image starts]: Múltiplos angiomiolipomas renais na ressonância nuclear magnética (RNM) coronal em T1Cortesia do Dr. Francis J. DiMario Jr [Citation ends].
[Figure caption and citation for the preceding image starts]: Múltiplos angiomiolipomas renais na ressonância nuclear magnética (RNM) coronal em T1Cortesia do Dr. Francis J. DiMario Jr [Citation ends].
linfangioleiomiomatose pulmonar
Afeta até 50% das mulheres com CET e parece ser hormonalmente sensível.[27]
As apresentações sintomáticas podem consistir em pneumotórax ou quilotórax espontâneo.
A linfangioleiomiomatose (LAM) produz cavitações císticas progressivas no interior dos pulmões, que causam uma diminuição na troca de ar e, por fim, óbito. O tratamento é de suporte até que possa ser realizado um transplante de pulmão.
Essa lesão pulmonar no CET parece ser causada por metástases de angiomiolipomas renais subjacentes. Também ocorre LAM esporadicamente em mulheres sem CET.[2][3][24][Figure caption and citation for the preceding image starts]: Lesões císticas na linfangioleiomiomatose (LAM) do pulmão em tomografia computadorizada (TC) axialCortesia do Dr. Francis J. DiMario Jr [Citation ends].
nódulos subependimários calcificados cerebrais
Nódulos subependimários são identificáveis nas áreas adjacentes à parede ventricular como pequenas protuberâncias para o interior da cavidade do líquido cefalorraquidiano.
Elas são frequentemente calcificadas na primeira infância, mas essa calcificação pode evoluir mais gradualmente ao longo da infância. A calcificação característica é facilmente identificável com tomografia computadorizada, mas também pode ser identificada com ressonância nuclear magnética.
Os nódulos são compostos de astrócitos displásicos e de componentes astrocíticos de linhagens mistas.[2][3][24][Figure caption and citation for the preceding image starts]: Nódulos subependimários calcificados na tomografia computadorizada (TC)Cortesia do Dr. Francis J. DiMario Jr [Citation ends].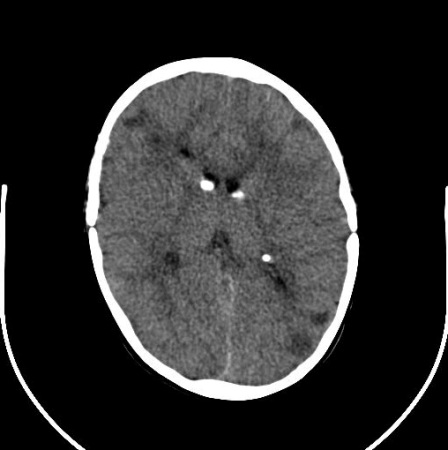 [Figure caption and citation for the preceding image starts]: Nódulos subependimários e túberos corticais em imagem axial de ressonância nuclear magnética (RNM) em T2Cortesia do Dr. Francis J. DiMario Jr [Citation ends].
[Figure caption and citation for the preceding image starts]: Nódulos subependimários e túberos corticais em imagem axial de ressonância nuclear magnética (RNM) em T2Cortesia do Dr. Francis J. DiMario Jr [Citation ends].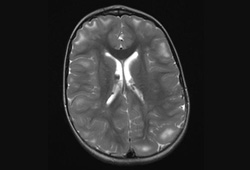
múltiplos tubérculos corticais e/ou linhas de migração radial
Regiões de displasia cortical focal estendendo-se para as áreas justacorticais da substância branca dando a aparência de lesões de massa giral. Elas podem ser identificadas como áreas de baixa densidade na tomografia computadorizada e áreas de alta intensidade nas sequências de ressonância nuclear magnética (RNM) com recuperação da inversão atenuada por fluidos (FLAIR)/T2.
As linhas de migração são vistas como trilhas lineares de lesões de alta intensidade de sinal em sequências de RNM em T2 que se estendem de forma centripetamente perpendicular às margens ventriculares que representam uma trilha de componentes celulares migrados de forma incompleta ao longo dos remanescentes da glia radial.
astrocitoma de células gigantes
Nódulos subependimários podem crescer em virtude de proliferação celular, com predileção pela região em torno do forame de Monro. Portanto, em virtude do tamanho e do local, eles são designados como um astrocitoma subependimário de células gigantes (ASCG).
Eles ocorrem em cerca de 10% a 15% dos pacientes, em geral até os 10 anos de idade.[28]
O crescimento pode produzir obstrução das vias do líquido cefalorraquidiano e invasão das regiões hipotalâmicas e quiasmáticas subjacentes. Não é possível predizer a taxa de crescimento e a necessidade iminente de intervenção neurocirúrgica. São necessários exames de imagem de acompanhamento e critérios clínicos.[2][3][24][Figure caption and citation for the preceding image starts]: Grande astrocitoma subependimário de células gigantes na ressonância nuclear magnética (A-axial T2)Cortesia do Dr. Francis J. DiMario Jr [Citation ends]. [Figure caption and citation for the preceding image starts]: Grande astrocitoma subependimário de células gigantes na ressonância nuclear magnética (B-sagital T1)Cortesia do Dr. Francis J. DiMario Jr [Citation ends].
[Figure caption and citation for the preceding image starts]: Grande astrocitoma subependimário de células gigantes na ressonância nuclear magnética (B-sagital T1)Cortesia do Dr. Francis J. DiMario Jr [Citation ends].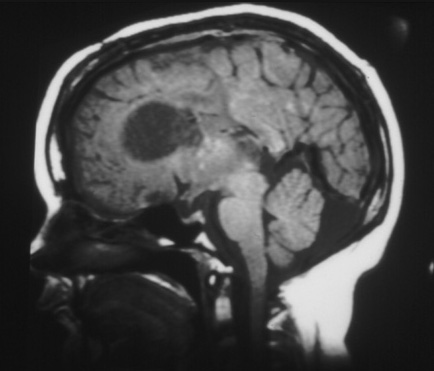
angiofibromas faciais
Esses nódulos hamartomatosos de tecido conjuntivo e vascular se distribuem em um padrão de borboleta. Mais comumente, eles surgem em torno dos 3 a 5 anos de idade, aumentam de tamanho e número ao longo da vida e são identificados em até 88% dos pacientes com CET.[29] Eles também podem ser vistos em cerca de 80% dos pacientes com NEM-1 (neoplasia endócrina múltipla).[30][Figure caption and citation for the preceding image starts]: Angiofibromas faciaisCortesia do Dr. Francis J. DiMario Jr [Citation ends].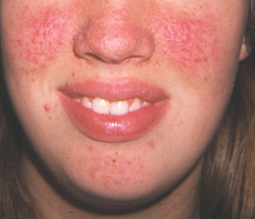
placa(s) cefálica(s)
Placas cefálicas são manchas hamartomatosas de tecido vascular e conjuntivo com deposição de colágeno.
Elas se localizam tipicamente na testa ou na linha do couro cabeludo, e estão geralmente presentes no nascimento e aumentam e se calcificam até a idade adulta.[Figure caption and citation for the preceding image starts]: Placa na testaCortesia do Dr. Francis J. DiMario Jr [Citation ends].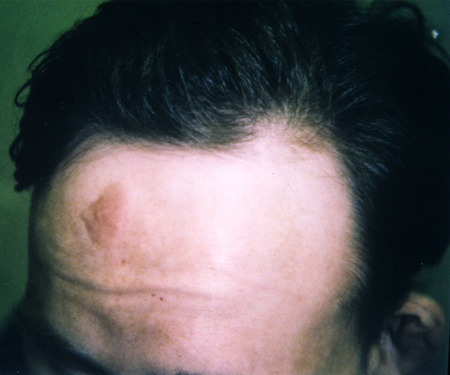
Eles são observados em cerca de 10% dos pacientes com CET.[2][3][31]
fibromas ungueais ou periungueais não traumáticos
Esses hamartomas de tecido conjuntivo podem ser múltiplos ou isolados.
Eles são encontrados mais frequentemente nos pododáctilos que nos dedos e mais no sexo feminino que no masculino.
Eles se desenvolvem geralmente na segunda década de vida.
Eles são observados em cerca de 20% dos pacientes com CET.[32]
Podem ser induzidos por trauma em pacientes sem CET.[2][3][31][Figure caption and citation for the preceding image starts]: Fibroma unguealCortesia do Dr. Francis J. DiMario Jr [Citation ends].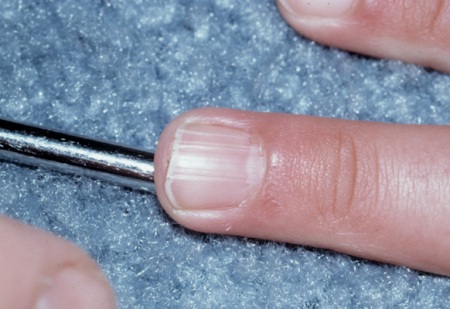
máculas hipomelanóticas
Três manchas são observadas em cerca de 90% dos pacientes.[33] As clássicas máculas hipomelanóticas (em formato de folha) assumem o formato piramidal característico, com base redonda e extremidade pontuda. Elas geralmente podem ser observadas a olho nu, mas a visualização pode ser melhorada com o uso de uma lâmpada de Wood.[Figure caption and citation for the preceding image starts]: Máculas hipomelanóticas e marcas de ShagreenCortesia do Dr. Francis J. DiMario Jr [Citation ends].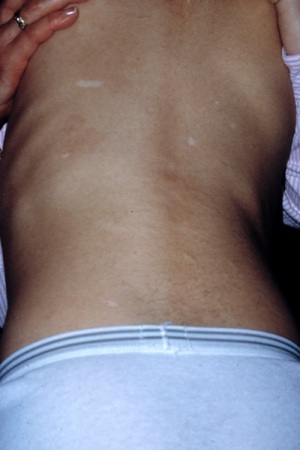
Estudos das áreas cutâneas identificaram função sudomotora anormal, com diminuição do volume de suor e inervação pós-ganglionar simpática anormal.[2][3][31]
marca(s) de Shagreen (nevo do tecido conjuntivo)
Localizadas, com mais frequência, no flanco lombossacral, mas também podem ser encontradas em outros locais.[Figure caption and citation for the preceding image starts]: Máculas hipomelanóticas e marcas de ShagreenCortesia do Dr. Francis J. DiMario Jr [Citation ends].
Geralmente são evidentes até os 10 anos de idade e podem ser dominantemente hereditárias sem CET.
hamartoma(s) nodular(es) retiniano(s)
Identificados por meio de fundoscopia sob midríase.
Compostos por fibras astrocíticas gliais ou por uma mancha acrômica, eles geralmente não causam perturbação visual e são evidentes até os 2 anos de idade. Eles podem evoluir de translúcidos a apresentar calcificação.
As lesões são observadas em cerca de 50% a 65% dos pacientes com CET, sendo bilaterais em 50% dos pacientes.[2][3][31]
[Figure caption and citation for the preceding image starts]: Hamartoma retinianoCortesia do Dr. Francis J. DiMario Jr [Citation ends].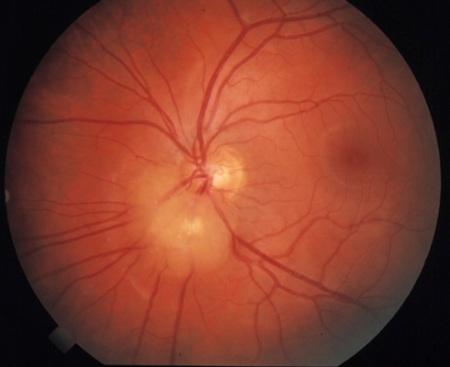
Incomuns
doença renal policística
A doença renal policística ´é infrequentemente encontrada em pacientes com CET (aproximadamente <5%).[25][Figure caption and citation for the preceding image starts]: Rins policísticosCortesia do Dr. Francis J. DiMario Jr [Citation ends].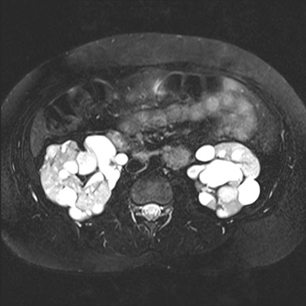 A probabilidade é maior para pacientes com TSC2, pois a doença resulta de uma deleção/mutação genética contígua do gene PKD1, que é imediatamente adjacente ao gene TSC2.
A probabilidade é maior para pacientes com TSC2, pois a doença resulta de uma deleção/mutação genética contígua do gene PKD1, que é imediatamente adjacente ao gene TSC2.
A suspeita surge pela palpação de rins aumentados no neonato e é confirmada por imagens renais.
As complicações durante o início da vida incluem hipertensão, dor no flanco, insuficiência renal e, menos frequentemente, hematúria.
Como as pessoas com rins policísticos apresentam um crescimento contínuo dos cistos, o parênquima renal funcional se torna progressivamente mais fino e disfuncional, até a ocorrência de insuficiência renal e uma necessidade de transplante.
A preservação da função renal, pela minimização do risco de infecções do trato urinário e do aparecimento de nefrolitíase, é fundamental.[2][3][24]
Outros fatores diagnósticos
comuns
numerosas depressões no esmalte dentário e fibromas intraorais
Praticamente todos os pacientes com CET apresentam nódulos nos dentes permanentes.[34] Esses nódulos frequentemente são orifícios relativamente grandes e numerosos ao longo da superfície do esmalte.
Eles não causam predisposição ao excesso de cáries, mas podem ser úteis no rastreamento de parentes de primeiro grau assintomáticos de pacientes com CET.
Entretanto, nódulos isolados podem ser observados em pessoas não afetadas, não sendo, portanto, específicos para CET.
Fibromas intraorais podem ser observados em até 70% dos adultos com CET.[34] Caso sejam grandes, pode ocorrer sangramento local e interrupção do alinhamento dentário normal. Depressões e fibromas intraorais juntos não são comuns em pacientes sem CET.[2]
autismo
O transtorno do espectro do autismo é observado em cerca de 40% a 50% dos pacientes.[6]
Em pacientes com CET e transtornos do espectro autista, existe uma prevalência de 75% de comprometimento cognitivo e uma prevalência de 75% a 100% de epilepsia concomitante. Em combinação, esse grupo de problemas clínicos é denominado transtornos neuropsiquiátricos associados ao CET.[2][3][7][36]
comprometimento cognitivo
Incomuns
múltiplos pólipos cólicos hamartomatosos
Esses pólipos raramente são identificados, mas podem estar presentes em muitos pacientes com CET. A prevalência verdadeira é desconhecida.
É necessária biópsia para confirmar a histologia hamartomatosa, que deve distinguir esses pólipos de pólipos adenomatosos observados em outras doenças.[35]
Fatores de risco
Fortes
predisposição genética
Aproximadamente um terço das pessoas com CET têm herança autossômica dominante, enquanto os dois terços restantes dos casos são devidos a mutações esporádicas.[4]
O uso deste conteúdo está sujeito ao nosso aviso legal