Abordaje
La anamnesis cuidadosa, la exploración física meticulosa y el cariotipo de leucocitos periféricos son esenciales para realizar un diagnóstico preciso del síndrome de Turner.
Antecedentes
Edad en el momento del diagnóstico
Una cantidad relativamente baja de pacientes se diagnostica en la primera infancia. La mayor proporción de detección tiene entre 10 y 16 años de edad, debido a una combinación de estatura baja marcada y retraso puberal. Menos del 10% de los casos se diagnostica antes del nacimiento y otro 20% se detecta en la infancia debido a la presencia de linfedema, cuello alado o defectos cardíacos congénitos. Otro 10% se diagnostica en la edad adulta, debido a la amenorrea secundaria. Los perfiles de determinación en Europa y EE. UU. son similares.[9][10]
[Figure caption and citation for the preceding image starts]: Datos de los perfiles de determinación del síndrome de Turner; 300 mujeres; edad diagnóstico, edad (en años) en el momento del diagnósticoDe la colección personal de la Dra. Carolyn Bondy, MS (National Institute of Child Health and Human Development natural history study 2001-2007; McCarthy K, et al. Expert Rev Endocrinol Metab. 2008;3:771-775 [Citation ends].

Anamnesis del nacimiento
Aproximadamente el 10% de las recién nacidas con síndrome de Turner tiene una cardiopatía congénita grave clínicamente manifiesta, como estenosis aórtica, coartación aórtica o hipoplasia de cavidades izquierdas. Sin embargo, muchas otras tienen defectos que son clínicamente silenciosos o sutiles, como válvula aórtica bicúspide o conexiones venosas pulmonares anómalas parciales.[23] El diagnóstico puede ser claro en el momento del nacimiento, lo que ocasionalmente se debe a la presencia de características cardíacas, linfedema (hinchazón en el dorso de los pies) o cuello alado.
Estatura baja
El crecimiento deficiente es, a menudo, el síntoma primario. Las niñas con síndrome de Turner son relativamente pequeñas desde la infancia y generalmente están por debajo del percentil 5 para estatura en las curvas de crecimiento específico para la edad y el sexo a los 10 años de edad. La estatura también se puede evaluar mediante los parámetros específicos para Turner. Magic Foundation: growth charts Opens in new window
Por lo tanto, se debe tomar la anamnesis de crecimiento de la niña enfocada en las causas potenciales de la estatura baja en la niñez. También se deben obtener antecedentes detallados de la estatura familiar (padres, hermanos). Las mediciones en serie pueden revelar un cruce hacia abajo de percentiles y sugerir una velocidad de crecimiento anormalmente baja.
Se debe medir la longitud en posición supina en los lactantes hasta los 2 años de edad y a partir de ese momento se medirán de pie. La estatura baja es proporcionada e involucra al torso y las extremidades inferiores por igual.
El crecimiento de un niño se relaciona considerablemente con su potencial genético. El déficit en el crecimiento se debe evaluar mediante la predicción de la estatura parental media para identificar el curso de crecimiento genético de la niña. La estatura parental media en una niña se calcula de la siguiente manera:
(estatura de la madre en centímetros + estatura del padre en centímetros)/2 - 6.4 cm [(estatura de la madre en pulgadas + estatura del padre en pulgadas)/2 - 2.5 pulgadas].
Retraso puberal
Aproximadamente el 15% de las niñas desarrolla tejido ovárico funcional y pubertad espontánea, pero la mayoría presenta retraso puberal y amenorrea primaria. Casi todas las jóvenes presentan menopausia prematura.[7]
Otras historias clínicas
Ataques frecuentes o inusualmente graves de otitis media en la infancia.
Habilidades sociales deficientes.
Exploración física
Un examen general puede revelar rasgos dismórficos patognomónicos:
[Figure caption and citation for the preceding image starts]: Características patognomónicas del síndrome de TurnerDe la colección personal de Carolyn Bondy, MS, MD (con datos de Ullrich O. Z. Kinderheilk. 1930;49:271-76 y Turner HH. Endocrinology. 1938;23:566-74) [Citation ends].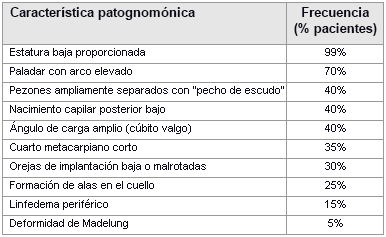
Otras características incluyen las siguientes:
Ojos o párpados caídos, ptosis.
Múltiples nevos melanocíticos.
Uñas hiperconvexas distróficas.
Escoliosis.
Estado puberal
Generalmente el vello púbico es disperso y el desarrollo puberal es mínimo.
Examen cardiovascular
Los soplos o chasquidos sugieren enfermedad de la válvula aórtica.
Se debe medir la presión arterial en todas las extremidades y palpar los pulsos femorales. La presencia de hipertensión en una o ambas extremidades superiores sugiere coartación aórtica.
La prevalencia de una cardiopatía congénita es mucho mayor en aquellas pacientes con evidencia clara de linfedema fetal, el cual se puede describir como inflamación de los tejidos, sobre todo de la cabeza y el cuello, debido a un desarrollo linfático disminuido. Algunas manifestaciones posnatales comunes son el cuello alado con nacimiento capilar y orejas de implantación baja.
A continuación, se muestran las comorbilidades/complicaciones que se observan con frecuencia en el síndrome de Turner.
[Figure caption and citation for the preceding image starts]: Asociación de los defectos cardiovasculares con el cuello aladoDatos compilados de Sachdev V, et al. J Am Coll Cardiol. 2008; 51:1904-1909, y Loscalzo ML, et al. Pediatrics. 2005;115:732-735 [Citation ends].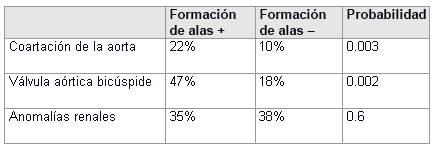 [Figure caption and citation for the preceding image starts]: Comorbilidades y complicaciones en niñas con síndrome de Turner; el diagnóstico de los defectos cardíacos congénitos se realizó mediante ecocardiograma y angiografía por resonancia magnética cardíaca; los estudios por imágenes renales y hepáticos se realizaron mediante ultrasonido; la hipertensión se determinó mediante monitorización ambulatoria de la presión arterial con estándares basados en la estatura; *incluye retorno venoso pulmonar anómalo parcial, arco aórtico transverso elongado, arco aórtico derecho; ** aumento de aspartato transaminasa (AST) o alanina transaminasa (ALT) >10%McCarthy K, et al. Expert Rev Endocrinol Metab. 2008;3:771-775 [Citation ends].
[Figure caption and citation for the preceding image starts]: Comorbilidades y complicaciones en niñas con síndrome de Turner; el diagnóstico de los defectos cardíacos congénitos se realizó mediante ecocardiograma y angiografía por resonancia magnética cardíaca; los estudios por imágenes renales y hepáticos se realizaron mediante ultrasonido; la hipertensión se determinó mediante monitorización ambulatoria de la presión arterial con estándares basados en la estatura; *incluye retorno venoso pulmonar anómalo parcial, arco aórtico transverso elongado, arco aórtico derecho; ** aumento de aspartato transaminasa (AST) o alanina transaminasa (ALT) >10%McCarthy K, et al. Expert Rev Endocrinol Metab. 2008;3:771-775 [Citation ends].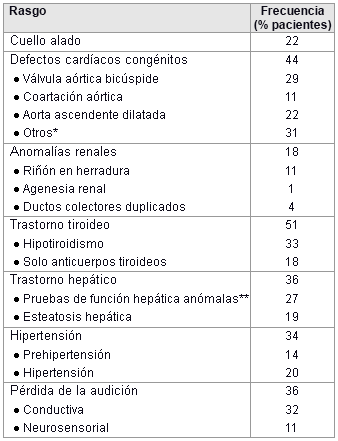
Cariotipo
El criterio de referencia para el diagnóstico durante las últimas 3 décadas ha sido el análisis citogenético de un cariotipo de sangre periférica; esta prueba es la base de la mayoría de las observaciones clínicas y la información de pronóstico. En los últimos años se ha avanzado en el uso de métodos de prueba molecular modernos mediante secuenciación de ADN o análisis de microarrays, pero aún no se encuentran ampliamente disponibles.[24][25]
Un cariotipo de leucocitos periféricos con 30 células examinadas es la prueba estándar para el diagnóstico.[26] Más del 10% de las células pierde todo o una parte significativa de un cromosoma sexual. Esta prueba identifica un mosaicismo del 10% o mayor, con una certeza del 95%. Se indica en los siguientes casos:[27]
Una mujer con una de las siguientes características clínicas:
Higroma quístico fetal o hidropesía, especialmente cuando es grave
Estatura baja idiopática
Características faciales distintivas
Defectos cardíacos congénitos obstructivos del lado izquierdo
Retraso inexplicable de la pubertad o la menarquia
Pareja con infertilidad inexplicable
Una mujer con al menos dos de las siguientes características clínicas:
Anomalía renal (herradura, ausencia o hipoplasia)
Deformidad de Madelung
Problemas neuropsicológicos o problemas psiquiátricos
Múltiples nevos típicos o melanocíticos
Uñas displásicas o hiperconvexas
Otros defectos cardíacos congénitos
Edad <40 años con discapacidad auditiva y baja estatura.
Se recomienda repetir la prueba en los siguientes casos:[5]
A los bebés diagnosticados prenatalmente se les debe realizar un cariotipo posnatal para confirmar el diagnóstico
Individuos diagnosticados únicamente mediante hisopo bucal
Individuos con un cariotipo realizado en un pasado lejano
Cuando el informe original no está disponible para su revisión.
Se envía una muestra de sangre de 15 mL, la cual se recolecta mediante una técnica estéril y se mantiene a temperatura ambiente, al laboratorio de genética dentro de las 24 horas. Según el American College of Medical Genetics, se debe contar con un mínimo de 20 células para determinar el número cromosómico cuando se busquen posibles anomalías cromosómicas sexuales en las que el mosaicismo sea común. El análisis está completo si se confirma el mosaicismo. Si se observa una célula con pérdida, ganancia o reordenación de cromosomas sexuales dentro de las primeras 20 células analizadas, deberán evaluarse un mínimo de 10 células adicionales.[28][29]
En pacientes con una sospecha de diagnóstico fuerte, si la prueba demuestra que <10% de las células son anómalas, deben contarse más metafases junto con una hibridación fluorescente in situ (FISH) o analizarse otros tipos de células con interfase FISH y verificarse con un genetista o citogenetista.[30] Según la citogenética, las pacientes con síndrome de Turner se clasifican de la siguiente manera:
45,X sin mosaicismo
Cromosoma X único en todas las células somáticas (monosomía X); comprende alrededor del 60% de las pacientes con síndrome de Turner.
X o Y fragmentado
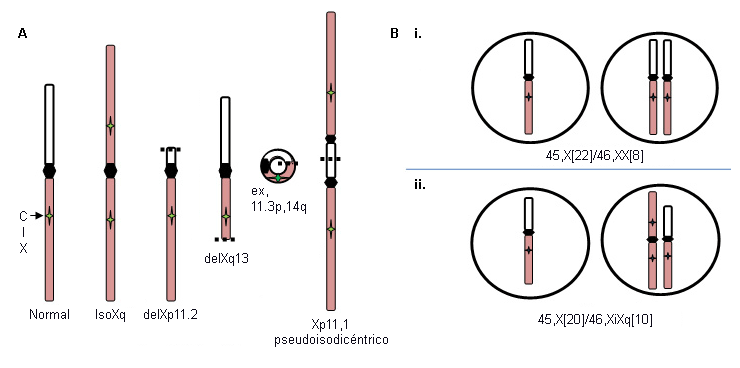
Deleciones Xp (46,X,delXp), cromosomas isoXq (46,X,iXq), deleciones Xq y cromosomas X o Y en anillo con deleciones intersticiales sustanciales. El cromosoma isoXq, que proviene de la deleción de Xp y la fusión de 2 brazos largos, es la anomalía estructural más común asociada al síndrome de Turner. La deleción de una gran parte de Xp también se asocia con el síndrome. A menudo, el cromosoma sexual anómalo se pierde en algunas células durante el desarrollo embrionario, lo que da como resultado el mosaicismo de una estirpe celular 45, X, además de la línea X frágil, 46, X (ver en la figura A a continuación).
La detección de un cromosoma pequeño en anillo o marcador requiere de análisis adicional mediante FISH con marcadores específicos para Y. Aquellos con un cromosoma Y evidente requieren de evaluación adicional debido a los riesgos de gonadoblastoma.
Mosaicismo 45,X
Existen dos tipos de mosaicismo.
La pérdida de un cromosoma sexual durante las divisiones celulares embrionarias tempranas puede provocar una combinación de células 46,XX normales y células 45,X en proporciones variables en los tejidos corporales e involucra a alrededor del 15% de las pacientes con síndrome de Turner: por ejemplo, 45,X (50%)/46,XX (50%) (ver en la figura B i. a continuación). La proporción relativa de células normales en diferentes tejidos influye en el fenotipo.
Por el contrario, la formación de un embrión 46,X,abnX suele ir acompañada de la pérdida del X fragmentado durante algunas divisiones celulares embrionarias, lo que da lugar a un mosaicismo para una línea celular monosómica, 45,X, junto con una línea celular que contiene un X fragmentado; por ejemplo, iXq (véase la figura B ii. a continuación). Este individuo no presenta células normales; se espera el fenotipo completo.
[Figure caption and citation for the preceding image starts]: Alteraciones del cromosoma X en el síndrome de Turner; ver explicación en el textoDe la colección personal de la Dra. Carolyn Bondy, MS [Citation ends].
Investigaciones posteriores
Edad ósea
El método de evaluación de la maduración esquelética es la radiografía. Por lo general, muestra un retraso leve (2 años menos que la edad cronológica). Se recomienda evaluar el potencial de crecimiento en todas las niñas prepúberes con el síndrome.
Hormona antimülleriana (AMH) y hormona foliculoestimulante (FSH) sérica[5]
Una FSH elevada y una AMH muy baja o no detectable pronostican una insuficiencia ovárica completa. Sin embargo, el potencial ovárico no se puede pronosticar con certeza en las niñas, ya que los niveles de FSH se elevan al rango menopáusico solamente en la edad de la pubertad normal en las mujeres con insuficiencia ovárica.
Ultrasonido pélvico
Identifica un útero inmaduro y una morfología pequeña de ovario rudimentario.
Se realiza en niñas mayores y mujeres.
Los ovarios se forman de manera normal en los fetos femeninos 45,X, aunque la mayoría presenta muerte acelerada del ovocito y degeneración ovárica en forma de estrías fibrosas.[21]
Examen del esqueleto
Se realiza en la infancia a los 5-6 años y a los 12-14 años para evaluar anomalías asociadas, como deformidades de la muñeca y escoliosis.[5]
La deformidad de Madelung (cúbito distal prominente) solamente se halla en alrededor del 5% de los pacientes, pero se dan con frecuencia grados menores de anomalías de la muñeca que pueden conducir a problemas funcionales.
Análisis de sangre iniciales para detectar comorbilidades/complicaciones[5]
Pruebas de función tiroidea (PFT) para enfermedad tiroidea autoinmune en todos los pacientes en el momento del diagnóstico. Pueden medirse los anticuerpos tiroideos si las PFT son anormales.
LFT para descartar la "hepatitis Turner" si tiene 10 años o más.
Glucosa en ayunas y HbA1c para identificar el riesgo de diabetes si tiene 10 años o más.
Lípidos séricos para evidencia de dislipidemia si tiene 18 años o más y si existe al menos un factor de riesgo de enfermedad cardiovascular (consulte también las recomendaciones regionales).
Nivel de IgA y transglutaminasa tisular IgA para detectar enfermedad celíaca si tiene 2 años.
Niveles de vitamina D controlados después de los 9 años.
Además, en el momento del diagnóstico debe realizarse lo siguiente:
Cribado para pérdida de la audición conductiva o neurosensitiva[5]
Se debe realizar una evaluación audiológica formal para garantizar medidas técnicas y de rehabilitación tempranas y adecuadas.
Detección de errores refractivos y alteraciones visuales[5]
Se han informado estrabismo, ambliopía, hipermetropía y miopía, ptosis y daltonismo. Las mujeres con síndrome de Turner tienen un pliegue epicántico (pliegue del párpado superior que cubre la esquina interna del ojo) e hipertelorismo (aumento de la distancia entre los ojos). Se debe realizar un examen oftalmológico completo entre los 12 y 18 meses o en el momento del diagnóstico, si es de mayor edad.
Cribado en busca de anomalías renales congénitas
Se debe realizar una ecografía renal para evaluar la presencia de anomalías estructurales, como riñón en herradura, agenesia renal y sistema colector duplicado, que afecta a aproximadamente el 25% de las pacientes con síndrome de Turner.[3][4][5] La función renal es generalmente normal, pero la obstrucción del sistema colector se asocia a infecciones urinarias y puede requerir corrección.
Cribado en busca de defectos cardiovasculares congénitos
Al momento del diagnóstico sin importar la edad, se debe realizar una evaluación cardiovascular integral por parte de un consultor en cardiopatías congénitas.[27]
Se debe intentar la visualización de la válvula aórtica, la aorta torácica y las venas pulmonares mediante ecocardiografía transtorácica (ETT) en bebés o mediante resonancia magnética cardíaca (RMC) o TC en niños mayores y adultos. La RMC requiere sedación en pacientes jóvenes y, por lo tanto, si las evaluaciones clínicas y ecocardiográficas parecen normales, es razonable esperar hasta que el niño tenga edad suficiente para cooperar sin sedación (generalmente entre 9 y 10 años) para realizar una RMC de detección.
Se debe realizar un ECG para evaluar posibles alteraciones de conducción y repolarización.
Si se encuentran defectos congénitos, el seguimiento y el tratamiento dependerán de cada afección individual.[27]Las niñas con defectos cardiovasculares deben pasar a una cirugía para cardiopatías congénitas en adultos, ya que se encuentran en riesgo constante de sufrir complicaciones aórticas durante la edad adulta.
[Figure caption and citation for the preceding image starts]: La imagen por resonancia magnética cardíaca revela un arco aórtico normal en forma de ”bastón de caramelo” a la izquierda, en comparación con una coartación aórtica sin diagnóstico previo, inmediatamente después del origen de la arteria subclavia izquierda (flecha), detectada mediante imagen por resonancia magnética en una mujer adulta con síndrome de Turner con hipertensión grave de la parte superior del cuerpoDe la colección personal de la Dra. Carolyn Bondy, MS (estudio realizado por National Institutes of Health [NIH]) [Citation ends].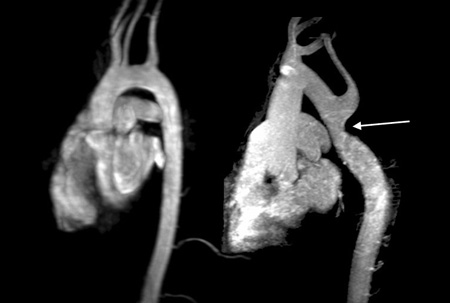
Las alteraciones más frecuentes son coartación aórtica y válvulas aórticas bicúspides. Otras alteraciones incluyen venas pulmonares anómalas parciales, hipoplasia de cavidades izquierdas y dilatación de la aorta. La prevalencia de defectos cardiovasculares es mucho mayor en pacientes con evidencia clara de linfedema fetal, tales como el cuello alado. En el 30% de las pacientes asintomáticas se observa una válvula bicúspide funcional como resultado de la fusión completa o parcial de los velos coronarios derecho e izquierdo, lo que implica un riesgo de infección, deterioro de las válvulas, dilatación y disección aórtica.[18] Los defectos cardiovasculares congénitos son la causa principal de mortalidad prematura en el síndrome de Turner. Dos genes, TIMP1 y TIMP3, que están ubicados en el brazo corto del cromosoma X cuando son hemicigotos, aumentan el riesgo de aortopatía con una probabilidad de 12.86.[19] En una angiografía por RM (ARM), la coartación aórtica se puede ver como un estrechamiento focal, a diferencia de la apariencia aórtica normal que es un arco aórtico parejo en forma de ”bastón de caramelo”.
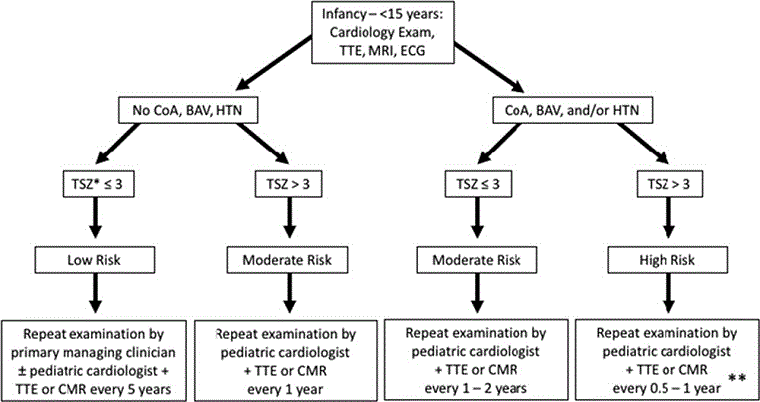
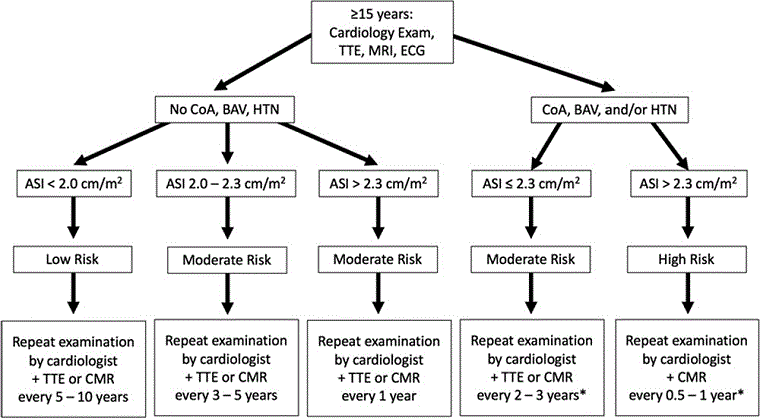
Los algoritmos basados en las guías de práctica clínica de la American Heart Association para la salud cardiovascular en el síndrome de Turner ayudarán en la investigación y el seguimiento.[27]
[Figure caption and citation for the preceding image starts]: Algoritmo para cribado y monitorización de la enfermedad cardiovascular congénita en el síndrome de Turner en niñas menores de 15 añosAdaptado de: Gravholt CH, Andersen NH, Conway GS, et al. Guías de práctica clínica para el cuidado de las niñas y las mujeres con síndrome de Turner: procedimientos de la reunión internacional contra el síndrome de Turner en Cincinnati 2016. Eur J Endocrinol. 2017;177(3):G1-G70. [Citation ends].
[Figure caption and citation for the preceding image starts]: Algoritmo para cribado y monitorización de la enfermedad cardiovascular congénita en el síndrome de Turner en mujeres y niñas mayores de 15 años. (ECC, enfermedad cardiovascular congénita; VA, válvula aórtica; VAB, válvula aórtica bicúspide; APVP, venas pulmonares anómalas parciales; Coarc, coartación; CPCA, cardiopatía congénita del adulto; ACV, aparato cardiovascular; HT, hipertensión)Adaptado de: Gravholt CH, Andersen NH, Conway GS, et al. Guías de práctica clínica para el cuidado de las niñas y las mujeres con síndrome de Turner: procedimientos de la reunión internacional contra el síndrome de Turner en Cincinnati 2016. Eur J Endocrinol. 2017;177(3):G1-G70. [Citation ends].
El uso de este contenido está sujeto a nuestra cláusula de exención de responsabilidad