Abordagem
É essencial obter uma história detalhada e realizar exames físicos e neurológicos completos em qualquer lactente que apresentar distonia. Isso geralmente permite que o médico classifique a distonia de acordo com as características clínicas, o que também sugere se a causa é hereditária ou adquirida. Investigações adicionais podem ser necessárias, para as quais a abordagem geral do autor é descrita na imagem a seguir.
[Figure caption and citation for the preceding image starts]: Distonia infantil: avaliação e diagnóstico genéticoCriado pelo BMJ Knowledge Centre [Citation ends].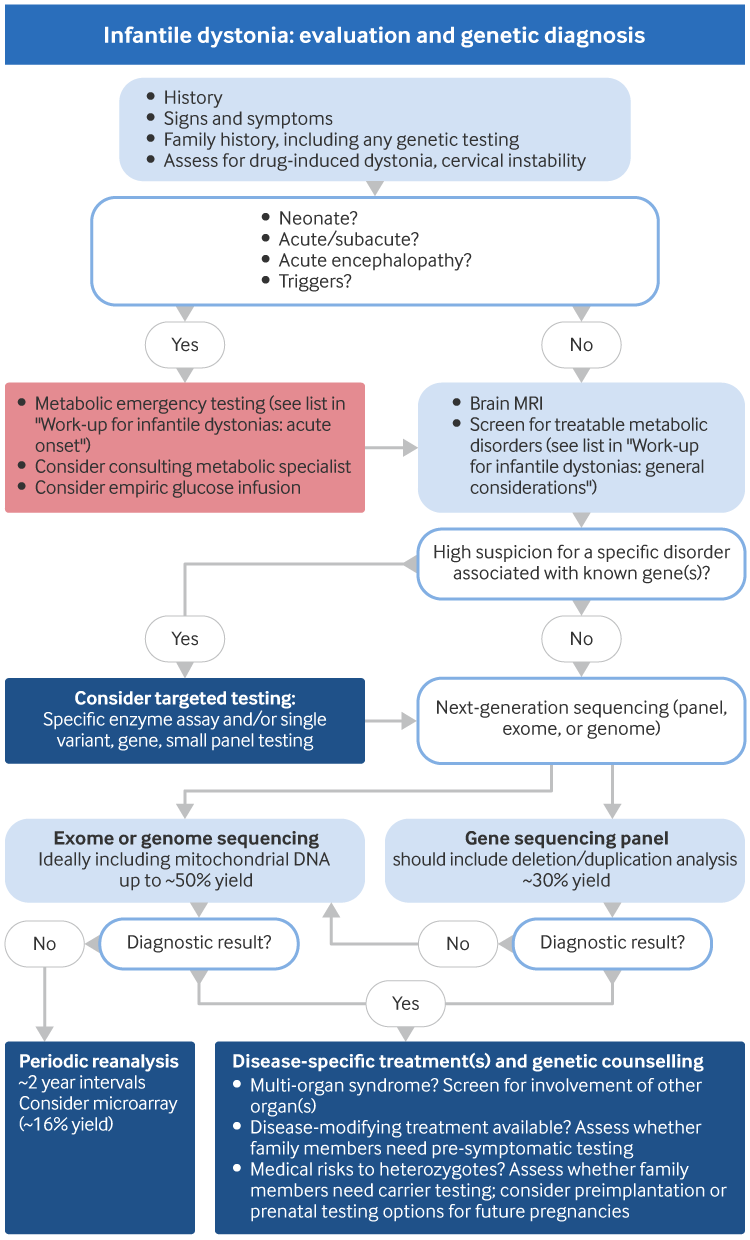
História
As informações essenciais que devem ser reunidas a partir dos pais incluem:
História familiar
Consanguinidade
História de exposição a medicamentos/toxinas
História de lesão perinatal
História de infecção do sistema nervoso central
História de trauma cranioencefálico/no pescoço
Idade de início
Parte do corpo inicialmente envolvida e evolução da distonia
Fatores desencadeantes (por exemplo, medicamentos, infecções, atividade, exercício); fatores agravantes (por exemplo, fatigabilidade) e variações diurnas; fatores atenuantes (por exemplo, sono)
Informações com relação à evolução da distonia (por exemplo, crônica, paroxística ou transitória).[114]
Os pais devem ser encorajados a fornecer fotos e a filmar seus filhos. Estas são ferramentas extremamente úteis na avaliação do lactente com movimentos intermitentes anormais.
Quadro clínico
O quadro clínico e as características históricas observadas ajudarão a distinguir a distonia infantil de outros distúrbios do movimento anormal. No entanto, pode ser difícil distinguir a distonia de outras causas de hipertonia em lactentes, como a rigidez e a espasticidade. Para crianças mais velhas, existe uma ferramenta de avaliação quantitativa da hipertonia.[79][115] Hypertonia assessment tool Opens in new window
Os movimentos distônicos são padronizados e sustentados. Eles envolvem repetidamente os mesmos grupos musculares e tipicamente causam torção de partes do corpo. A distonia geralmente piora com fadiga e estresse emocional, e melhora com sono e relaxamento.[4][116][117][118] Os movimentos passivos são resistentes à velocidade de movimento dos membros muito lenta (ao contrário da espasticidade, que é dependente da velocidade). Gestos antagonistas (truques sensoriais) são bem descritos na distonia focal que afeta o pescoço, mãos e face. A melhora nos movimentos distônicos é normalmente mantida enquanto o truque é realizado pelos pacientes, mas desaparece quando o gesto termina.[7]
Outros distúrbios do movimento anormal diferenciais têm as seguintes características.
A rigidez é uma resistência elevada a movimentos passivos ou articulações, que é uniforme independentemente da velocidade e direção do movimento (flexão ou extensão), tradicionalmente comparada à resistência de um "cano de chumbo". A resistência pode ser interrompida ritmicamente, resultando no fenômeno de rigidez em roda dentada. A rigidez é mais perceptível com movimentos lentos.[5]
Na espasticidade, a hipertonicidade predomina nos músculos flexores e pronadores dos membros superiores, e a resistência ocorre no início do movimento e depois aumenta com a velocidade do movimento passivo. Esse é o fenômeno do canivete.
Os tiques são movimentos repetitivos dos músculos esqueléticos ou faringolaríngeos; estes são responsáveis pela emissão de sons ou ruídos. Eles são movimentos estereotipados, súbitos, inoportunos, não proposicionais, absurdos, irresistíveis e de intensidade variável. O paciente pode reproduzir tiques ou controlá-los voluntariamente. Os tiques podem persistir durante o sono.
O tremor é uma oscilação rítmica de uma parte do corpo ao redor de um ponto ou local fixo. Isso resulta em contrações sincrônicas alternadas de músculos antagonistas reciprocamente inervados. A ritmicidade é a característica mais útil na identificação do tremor.
A ataxia é um distúrbio do controle fino da postura e do movimento. A característica inicial e mais proeminente geralmente é uma marcha anormal.
Nas distonias hereditárias e idiopáticas sem degeneração, a distonia e/ou outros movimentos anormais geralmente predominam. Cada tipo individual de distonia hereditária isolada tem achados específicos que fornecem indícios importantes para o diagnóstico exato. Algumas características importantes que podem sugerir uma síndrome de distonia particular situam-se no eixo das características clínicas da classificação da distonia. Além da idade de início e se existem outras características motoras (distonias isoladas, combinadas, complexas), elas incluem a distribuição da distonia e seu padrão temporal, incluindo se a evolução é estática ou progressiva, e qualquer variabilidade como a variação diurna ou ocorrência paroxística ou induzida por ação. Existe alguma sobreposição fenotípica entre doenças de diferentes genes da distonia devido à variabilidade na apresentação clínica: por exemplo, algumas distonias combinadas podem inicialmente se apresentar com fenomenologia aparentemente isolada.
Nas distonias adquiridas e nas distonias complexas hereditárias, a presença de outras anormalidades neurológicas e/ou sistêmicas sugere uma série de lesões. Elas incluem síndromes vasculares, infecções, síndromes neoplásicas, traumáticas e síndromes com doenças metabólicas e doenças degenerativas subjacentes.[114] Geralmente há deterioração neurológica progressiva na distonia sindrômica com doenças metabólicas subjacentes e doenças degenerativas. Assim como na distonia isolada hereditária, indícios importantes na história e no exame físico podem levar o médico ao diagnóstico do tipo específico de distonia.
Uma história familiar de distonia positiva pode sugerir uma etiologia hereditária. No entanto, uma história familiar negativa não exclui essa possibilidade, já que existem distonias onde são observadas herança autossômica recessiva, penetrância incompleta ou expressividade variável (como apenas 30% de penetrância na DYT-TOR1A) ou variantes patogênicas de novo para condições autossômicas dominantes.
O acompanhamento periódico dos lactentes com distonia geralmente é um pré-requisito para a elaboração de um diagnóstico preciso.
Distúrbios que mimetizam a distonia e armadilhas no diagnóstico
Estereotipias primárias, desvio tônico paroxístico benigno dos olhos na infância, torcicolo infantil secundário e convulsões devem ser considerados no diagnóstico diferencial da distonia.
A distonia dopa-responsiva (síndrome de Segawa, distonia-parkinsonismo com variação diurna) é comumente diagnosticada erroneamente como paralisia cerebral diplégica ou quadriplégica espástica, epilepsia intratável ou paraplegia espástica hereditária.[9][10][119] Ela deve ser considerada como um possível diagnóstico em qualquer criança com hipertonia paroxística progressiva de origem desconhecida.
A hiperecplexia hereditária infantil deve ser considerada quando se observa que um neonato tem rigidez generalizada. Os sinais característicos são uma resposta exagerada de sobressalto induzida por um estalo ou toque no nariz, os quais produzem uma rigidez flexora violenta e breve e apneia voluntária.[113]
A possibilidade de uma etiologia genética onde o tratamento oportuno e específico da doença poderia alterar significativamente o desfecho deve ser levada em consideração. Muitos distúrbios do movimento raros possuem hoje tratamentos conhecidos que podem mitigar, prevenir ou mesmo reverter os sintomas, resumidos recentemente pela Parkinson’s Disease Movement Disorders Society Task Force on Rare Movement Disorders.[120] A distonia dopa-responsiva (tratável com levodopa) deve ser considerada em qualquer paciente com diagnóstico desconhecido e ressonância nuclear magnética (RNM) normal. Em pacientes com distonia, convulsões e encefalopatia, estudos do líquido cefalorraquidiano (LCR) da tetraidrobiopterina e dos metabólitos de neurotransmissores podem detectar distúrbios dos neurotransmissores com tratamentos específicos (por exemplo, levodopa, tetraidrobiopterina, ácido folínico) com base na enzima afetada. Entre as possíveis etiologias da distonia de início agudo ou subagudo com encefalopatia estão as acidemias orgânicas com metabólitos tóxicos (onde o controle metabólico agudo ou crônico pode alterar o desfecho) e a doença dos núcleos da base responsiva a biotina-tiamina. A distonia paroxística com ou sem epilepsia pode ser uma consequência da deficiência de transportador 1 da glicose (GLUT1; SLC2A1), tratada com uma dieta cetogênica. As causas raras de distonia com neurodegeneração incluem deficiência de creatina cerebral e deficiência do transporte de folato cerebral, que podem responder a tratamento alimentar. Os achados da RNM podem sugerir deposição de manganês (hiperintensidade T1 palidal) nas hipermanganesemias, tratável com quelação.
Investigação para distonias infantis
Início agudo
Se a distonia for de início agudo ou parte de uma deterioração neurológica aguda, uma avaliação imediata é indicada para "emergência metabólica", bem como para outras situações urgentes listadas acima, como intoxicação por medicamentos ou encefalopatia bilirrubínica. A suspeita clínica é particularmente alta nos lactentes que já tiverem estado bem e depois piorarem progressivamente em um período de horas a dias; com nova encefalopatia ou convulsões; onde a história inclui possíveis gatilhos para descompensação metabólica, como doença febril, vômito ou jejum; e no período neonatal. O objetivo do exame metabólico é identificar distúrbios com intoxicação por moléculas pequenas (por exemplo, bilirrubina, amônia, ácidos orgânicos nas acidemias orgânicas) e tratá-los antes que evoluam para edema ou necrose cerebral. Um guia para exames de emergência está disponível no Vademecum Metabolicum. Vademecum Metabolicum Opens in new window Os exames incluem:
Gasometria arterial (ABG) e eletrólitos sanguíneos (avaliar para acidose e calcular o anion gap)
Glicose sanguínea
Amônia sérica
Lactato sérico
Ácido úrico sanguíneo
Transaminases hepáticas, estudos da coagulação
Creatina quinase sanguínea, proteína C reativa, creatinina
Cetonúria, glicose, proteína (tira reagente).
Além disso, testes de bilirrubina (para avaliação de kernicterus) e teste de sulfito na urina para deficiência de sulfito oxidase (se disponível) podem ser indicados nos neonatos. Amostras de sangue e urina também podem ser coletadas para exames metabólicos adicionais, como ácidos orgânicos na urina, conforme descrito abaixo, porque o rendimento é maior durante a doença aguda.
Enquanto as avaliações acima estiverem em andamento, o tratamento empírico pode ser considerado: infusão intravenosa de glicose a 10% em eletrólitos apropriados, com monitoramento do lactato e do pH sanguíneos. O objetivo da infusão de glicose é reduzir o catabolismo e, assim, diminuir a produção do composto neurotóxico, por isso é administrada mesmo que o paciente não esteja hipoglicêmico. O manejo adicional seria idealmente realizado em consulta com um especialista em metabolismo.
Considerações gerais
A primeira prioridade nos testes diagnósticos é avaliar as causas adquiridas e metabólicas que têm tratamentos específicos. A ressonância magnética do cérebro e os exames laboratoriais metabólicos são frequentemente necessários como parte disso. Testes genéticos moleculares podem ajudar a estabelecer o diagnóstico. Para os distúrbios do movimento pediátricos em geral, estão disponíveis abordagens diagnósticas publicadas com base na opinião de especialistas.[121] Na ausência de diretrizes consensuais específicas para avaliação da distonia infantil, a imagem acima mostra a abordagem geral dos autores.
Uma ressonância nuclear magnética (RNM) cranioencefálica é realizada em todos os lactentes e crianças em que a história e o quadro clínico sugerirem que a etiologia pode ser adquirida, lesional ou um distúrbio degenerativo. Na prática, mesmo se uma distonia primária for considerada como o diagnóstico mais provável, uma RNM cranioencefálica geralmente é realizada para descartar causas secundárias e avaliar os padrões das lesões ou a atrofia. Se a espectroscopia por ressonância magnética estiver disponível, ela deve ser realizada para avaliar os padrões associados a algumas doenças metabólicas, como a creatinina baixa nos distúrbios de deficiência de creatina cerebral que têm tratamentos específicos ou o lactato elevado nos distúrbios mitocondriais. A RNM é normal nas distonias isoladas, mas é frequentemente anormal nas distonias adquiridas ou degenerativas.
Em um paciente com início agudo ou subagudo dos sintomas em decorrência de etiologias de intoxicação, tais como acidemias orgânicas, a RNM pode mostrar lesões edematosas ou necróticas dos gânglios da base; um pico elevado de lactato pode sugerir um distúrbio mitocondrial ou uma doença dos gânglios da base responsiva à biotina-tiamina. A RNM em casos de doença crônica pode mostrar atrofia, malformação cerebral ou achados mais específicos, tais como depósito de ferro na neurodegeneração com acúmulo cerebral de ferro ou comprometimento da substância branca em distúrbios hipomielinizantes. A tomografia computadorizada cranioencefálica pode ser indicada para visualizar calcificações.
A escolha de quaisquer investigações adicionais será influenciada pelo quadro clínico e pelas possíveis causas secundárias que estão sendo consideradas.
Deve-se considerar o monitoramento por eletroencefalograma (EEG) ou por vídeo-EEG se a história sugerir convulsão, especialmente para doenças paroxísticas, como a distonia idiopática transitória da infância, o torcicolo paroxístico benigno da infância e as distonias adquiridas.
O exame metabólico pode ser considerado em todos os lactentes com distonia para avaliar os distúrbios com tratamentos específicos. Ele é particularmente importante quando a RNM cranioencefálica é anormal e as características clínicas sugerem uma causa metabólica. Às vezes ele é realizado no mesmo estágio da RNM cranioencefálica. Os exames laboratoriais seguintes fazem parte da investigação metabólica:
Análise da gasometria arterial
Amônia sérica
Análise de cetonas na urina
Análise de aminoácidos séricos
Rastreamento de ácidos orgânicos urinários para acidúrias orgânicas
Análise de ácido úrico sérico para a síndrome de Lesch-Nyhan
Teste de piruvato e lactato no soro ou no líquido cefalorraquidiano (LCR) para encefalomiopatias mitocondriais
Glicose sérica e no LCR para transportador de glicose tipo 1 (GLUT1)
estudos dos neurotransmissores do LCR[28]
Se a RNM cranioencefálica for normal e o diagnóstico não tiver sido estabelecido por exames prévios, deve-se considerar um ensaio terapêutico com levodopa. Esse teste é justificado em todos os pacientes com distonia de início precoce, para os quais o diagnóstico ainda não foi estabelecido.[122] Os sintomas melhoram consideravelmente na forma mais comum de distonia dopa-responsiva (síndrome de Segawa, distonia-parkinsonismo com variação diurna) em comparação com uma resposta lenta ou parcial em outros tipos de distonia.
A variação patogênica no gene da enxaqueca hemiplégica familiar (FHM1), CACNA1A, foi relatada em uma família com torcicolo paroxístico benigno no pai e no filho.[123] O teste é indicado se houver história familiar de torcicolo paroxístico benigno da infância ou um distúrbio similar, como enxaqueca hemiplégica.
Respostas automáticas do tronco encefálico, respostas auditivas evocadas do tronco encefálico e avaliação audiológica completa devem ser realizadas se o kernicterus ou as encefalomiopatias mitocondriais forem o diagnóstico preliminar como causa da distonia.
A atividade enzimática da hexosaminidase A e da hexosaminidase total pode ser estudada nos leucócitos ou em outros tecidos se a gangliosidose GM2 infantil for altamente considerada no diagnóstico diferencial da distonia.
Deve-se considerar uma biópsia muscular nos pacientes com distonia com possíveis encefalomiopatias mitocondriais se os estudos específicos de sequenciamento ou deleção/duplicação do genoma mitocondrial ou de genes codificados no núcleo não forem informativos.
Os testes genéticos moleculares são úteis, juntamente com testes bioquímicos/análises enzimáticas, se houver suspeita de uma deficiência enzimática específica.[124]
Teste genético molecular
Os testes genéticos relataram rendimentos diagnósticos de 30% a 60% em crianças com distonia, uma taxa mais alta do que na distonia em geral (aproximadamente 12% em coortes que incluem adultos).[2][125] O início precoce, a distonia combinada ou complexa e a ocorrência familiar aumentam a probabilidade de um diagnóstico genético.[2][125] Para atingir esses altos rendimentos, é importante escolher uma estratégia de testagem genética que leve em consideração o cenário clínico do paciente e os tipos de variantes que cada teste pode detectar.[126]
Na maioria dos pacientes, a variante genética patogênica é uma pequena variante de sequência (uma alteração de um único nucleotídeo ou inserção/deleção de alguns nucleotídeos) detectável por sequenciamento. Uma minoria de pacientes tem variantes do número de cópias (CNVs), como deleções ou duplicações; estes podem envolver um ou mais éxons dentro do gene (CNVs intragênicas), o gene inteiro ou múltiplos genes (microdeleções/duplicações).
Os testes genéticos comumente solicitados são os seguintes.
Testes de variantes específicas e testes de genes únicos: testes específicos estão disponíveis para algumas variantes comuns, como uma deleção específica de 3 nucleotídeos em muitos pacientes com DYT-TOR1A. Como esses testes são projetados para detectar apenas variantes específicas, os resultados negativos não descartam outras variantes no mesmo gene. Se um único gene precisar ser avaliado, isso pode ser feito usando-se uma combinação de sequenciamento de Sanger para identificar pequenas variantes de sequência e matriz no nível do éxon para identificar CNVs intragênicas.
Painéis genéticos: avaliam um subconjunto de genes associados à distonia, geralmente por meio de métodos de sequenciamento de última geração (NGS). Cada laboratório seleciona quais genes analisar. Os painéis genéticos quase nunca cobrem todos os genes relevantes, pois a distonia pode ocorrer em pacientes com síndromes como encefalopatias epilépticas e do desenvolvimento que geralmente não são consideradas distúrbios do movimento.[127] Os painéis genéticos detectam pequenas variantes de sequência. De preferência, eles também incluirão a detecção de CNVs intragênicas por métodos como matriz de éxon ou análise adicional de dados NGS; em caso afirmativo, o laboratório de testes declarará explicitamente que serão realizados testes de exclusão/duplicação (del/dup) ou testes de CNV. Mesmo com esses métodos, os painéis genéticos podem não detectar CNVs maiores e microdeleções/duplicações que abranjam todo o gene. O rendimento diagnóstico depende do número de genes analisados; um painel de 102 genes apresentou uma taxa de diagnósticos de 28% na distonia pediátrica (34% na distonia combinada e 16% na distonia isolada).[128]
Sequenciamento de exoma e genoma: esses métodos NGS avaliam éxons e sequências adjacentes (sequenciamento de exoma) ou sequências de todo o genoma (sequenciamento de genoma). O sequenciamento do DNA mitocondrial não é necessariamente incluído, mas geralmente é útil porque o diagnóstico diferencial pode incluir variantes do DNA mitocondrial, como as que causam a síndrome de Leigh. O sequenciamento do exoma e do genoma detecta pequenas variantes. Às vezes, eles também podem detectar CNVs com vários graus de sucesso.[129] O rendimento do sequenciamento do exoma foi de 60% a 66% em pequenas subcoortes de pacientes com distonia de início na infância.[2][3] Mais geralmente na distonia de início pediátrico, foram relatados rendimentos de 34% a 53%.[2][3] O sequenciamento do genoma tem um rendimento maior do que o sequenciamento do exoma na distonia.[125] Isso se deve principalmente à melhor qualidade dos dados de sequenciamento (permitindo maior sensibilidade para pequenas variantes) e melhor identificação de CNVs (que representaram 23% dos diagnósticos genéticos na coorte referenciada).[125]
Análise cromossômica por microarray: esses testes detectam principalmente grandes CNVs, como microdeleções/duplicações. Eles não detectam pequenas variantes de sequências. O menor tamanho de CNV detectável (resolução) varia entre os testes, mas geralmente não é suficiente para detectar CNVs intragênicas. Em um estudo, 16% dos pacientes com discinesia pediátrica apresentaram microdeleções de genes conhecidos da discinesia suficientemente grandes para detectar em microarray, com outros 12% com microdeleções possivelmente relevantes no microarray.[130]
Não há uma única escolha correta de qual teste solicitar primeiro, mas a decisão pode ser guiada pela amplitude do diagnóstico diferencial. Como os espectros fenotípicos de muitos genes da distonia se sobrepõem, muitas vezes é apropriado realizar um teste NGS avaliando múltiplos genes – painel de genes, exoma ou sequenciamento do genoma – como o teste genético inicial. As seguintes considerações se aplicam.
Em geral, o sequenciamento do exoma ou do genoma oferece as maiores taxas de diagnóstico, especialmente quando o diagnóstico diferencial é amplo. É apropriado solicitar o sequenciamento do exoma ou do genoma como teste inicial. Um painel de genes não precisa ser obtido primeiro.[131] Se o sequenciamento do exoma ou do genoma for solicitado, o laboratório deve receber informações clínicas detalhadas, incluindo características neurológicas, características não neurológicas, histórico familiar, qualquer teste genético anterior e quaisquer distúrbios/genes específicos que no topo do diagnóstico diferencial. O laboratório de testes genéticos pode usar essas informações para melhorar a qualidade da interpretação dos dados.
Quando há alta suspeita de um distúrbio específico, o teste de gene único ou painel de genes pode ter vantagens sobre o sequenciamento do exoma/genoma. Além do custo mais alto, o sequenciamento do exoma e do genoma apresenta um risco maior de retornar resultados indesejados, como variantes de significado incerto e achados secundários (variantes da doença em genes não relacionados à distonia). Testar menos genes reduz esse risco. Ocasionalmente, também é possível que o sequenciamento do exoma forneça um resultado falso-negativo: por exemplo, se a região que contém a variante do paciente tiver dados insuficientes para um resultado acurado.[132] Isso é um problema menor com o sequenciamento do genoma. Se for solicitado um painel genético, é preferível incluir a detecção de deleções/duplicações.
A análise cromossômica por microarray detecta grandes CNVs, que são um conjunto diferente das variantes pequenas/intragênicas encontradas principalmente nos testes NGS. Portanto, pode ser apropriado solicitar a análise por microarray juntamente com os outros testes. Se considerações práticas limitarem o número de testes que podem ser feitos, pode ser preferível solicitar um dos testes baseados em NGS, que têm rendimentos mais altos, e reservar o microarray para depois.
Se o teste do painel genético não for diagnóstico, convém passar para o sequenciamento de exoma ou genoma. O sequenciamento do exoma demonstrou aumentar a taxa de diagnósticos em pacientes que já apresentavam painéis genéticos negativos.[2]
Se o sequenciamento do exoma ou do genoma não for diagnóstico (particularmente com uma variante de significado incerto), é razoável solicitar uma reanálise do resultado do paciente aproximadamente a cada 2 anos.[133] Reanalisar a sequência do paciente à luz de novas informações sobre genes e variantes de doenças pode, às vezes, permitir que seu resultado seja revisado para uma classificação clinicamente mais útil, como causador de doença ou benigno.
Se for feito um diagnóstico genético, o tratamento e o aconselhamento específicos para a doença podem ser fornecidos com base nas características conhecidas da doença.
Se for diagnosticada uma síndrome de distonia complexa, o paciente pode ser rastreado quanto a quaisquer manifestações sistêmicas da doença.
Para condições em que o tratamento modifica o curso da doença, a consulta genética pode ser apropriada para determinar se algum membro da família deve ser submetido a testes pré-sintomáticos.
Em algumas condições, há riscos clínicos para os heterozigotos que justificam a avaliação dos pais ou de outros membros da família. Por exemplo, a mãe de um paciente com uma condição ligada ao cromossomo X pode estar em risco de sintomas de início tardio. O mesmo se aplica a um pai portador da mesma variante da doença que o probando em uma condição autossômica dominante com expressividade variável.
Conhecer a variante da doença no paciente permite testes direcionados para essa variante específica em outros membros da família. Os pais podem receber aconselhamento genético para discutir suas opções de testagem do portador, testagem pré-implantação e/ou teste pré-natal em gestações futuras.
O uso deste conteúdo está sujeito ao nosso aviso legal