Abordagem
Os pacientes com FC clássica geralmente apresentam retardo do crescimento pôndero-estatural no período neonatal ou na primeira infância. Alguns bebês e crianças mais novas apresentam bronquiolite prolongada ou grave e queixas respiratórias recorrentes. Adultos com FC não diagnosticados, que geralmente são pancreático-suficientes, podem apresentar bronquite, rinossinusite ou pancreatite crônica ou recorrente.
A FC é diagnosticada quando uma pessoa tem uma apresentação clínica de FC e evidências de disfunção do CFTR.[29] Quando há serviços de rastreamento de neonatos (por exemplo, como ocorre nos EUA e no Reino Unido), os casos serão diagnosticados imediatamente após o nascimento.
História
A história no nascimento deve incluir perguntas sobre a passagem do mecônio (qualquer atraso) e o local do nascimento (os hospitais dos EUA realizam o rastreamento de neonatos).História familiar positiva ou suspeita eleva o nível de suspeita para FC.
Trato respiratório
Pergunte à família sobre a frequência respiratória, retrações, tosse (quantidade e qualidade, inclusive a produção de escarro) e sibilos.Uma tosse produtiva, particularmente em episódios de tosse intensa, pode indicar FC.
Os pacientes podem apresentar infecções das vias aéreas inferiores recorrentes (bronquite ou pneumonia) que requerem antibióticos.Normalmente, as exacerbações pulmonares apresentam escarro raiado de sangue, mas alguns pacientes podem apresentar hemoptise maior (>300 cc/24 horas).
Trato gastrointestinal
Pergunte aos pacientes ou parentes sobre o apetite, alimentos típicos consumidos, hábitos intestinais (inclusive quantidade e qualidade) e qualquer refluxo gastroesofágico.
Um apetite insaciável, com fezes volumosas e gordurosas frequentes, é consistente com má absorção de gorduras e calorias.
Uma história de diminuição do número de fezes ao longo do tempo, com ou sem distensão abdominal ou vômitos, pode indicar obstrução intestinal.Ela é mais comum nos indivíduos com FC que naqueles sem FC.
Suspeita de insuficiência pancreática exócrina em pacientes com FC. Geralmente presente no nascimento ou na primeira infância.[30]
Os pacientes podem apresentar pancreatite e/ou apendicite aguda recorrentes.A presença de um defeito na condutância seletiva do bicarbonato pelo CFTR (exceto p.R75Q) aumenta o risco de pancreatite crônica em duas a quatro vezes.[31]
Exame físico
O exame físico pode estar normal em um paciente com FC.No entanto, alguns achados importantes devem elevar o nível de suspeita.
Desnutrição, manifestada como ausência de depósitos adiposos subcutâneos, abdome protuberante e peso abaixo do normal para a altura em lactentes e crianças pequenas.IMC nas crianças maiores, adolescentes e adultos.
Pólipos nasais.Consulte Pólipos nasais.
Diâmetro anteroposterior elevado do tórax e estertores na ausculta.
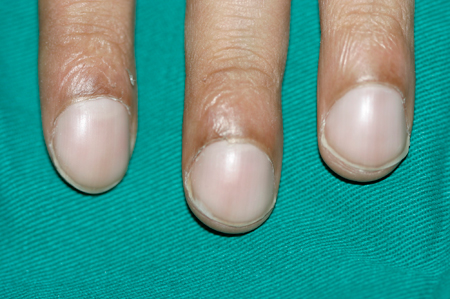
Baqueteamento digital das mãos.Consulte Avaliação do baqueteamento digital.
Massa fecal (geralmente no quadrante inferior direito), ou um fígado e/ou baço aumentados, ou ambos.Revelados por palpação abdominal.
Ausência bilateral do canal deferente, nos homens.
[Figure caption and citation for the preceding image starts]: Baqueteamento digitalDo acervo de Dr. Murlidhar Rajagopalan [Citation ends].
 [Figure caption and citation for the preceding image starts]: TC – Visão coronal dos seios paranasais mostrando o bloqueio do complexo ostiomeatal (Círculo Verde)Mohd Slim MA et al. Paediatric nasal polyps in cystic fibrosis. BMJ Case Rep 2016 Jun 21;2016; usado com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: TC – Visão coronal dos seios paranasais mostrando o bloqueio do complexo ostiomeatal (Círculo Verde)Mohd Slim MA et al. Paediatric nasal polyps in cystic fibrosis. BMJ Case Rep 2016 Jun 21;2016; usado com permissão [Citation ends].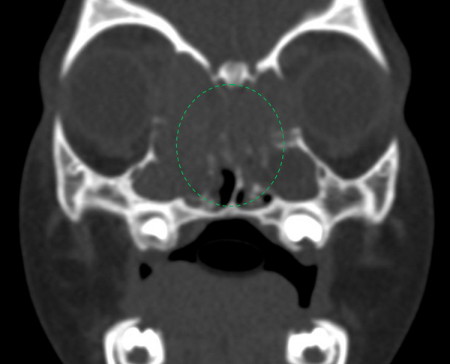 [Figure caption and citation for the preceding image starts]: Massa de tecido mole de cor pálida na cavidade nasal direitaMohd Slim MA et al. Paediatric nasal polyps in cystic fibrosis. BMJ Case Rep. 2016 Jun 21;2016; usado com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Massa de tecido mole de cor pálida na cavidade nasal direitaMohd Slim MA et al. Paediatric nasal polyps in cystic fibrosis. BMJ Case Rep. 2016 Jun 21;2016; usado com permissão [Citation ends].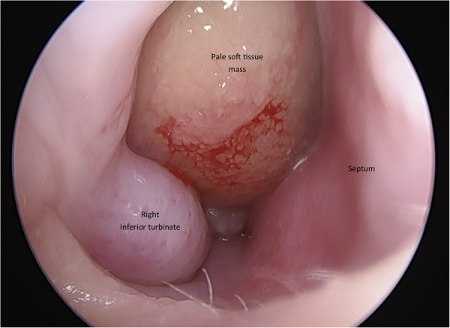
Investigações
As orientações da Cystic Fibrosis Foundation recomendam que os testes devem ser idealmente realizados de maneira hierárquica para estabelecer o diagnóstico: testes do suor primeiro, seguidos por testes genéticos do CFTR e, em seguida, testes fisiológicos do CFTR.Todas as pessoas diagnosticadas com FC devem fazer pelo menos um teste do suor e realizar uma análise genética do CFTR.
Teste do suor
Se houver suspeita (isto é, o paciente apresentar sintomas/sinais ou uma história familiar positiva), deve-se realizar o teste do suor (teste de iontoforese de pilocarpina).
Teste de suor negativo: uma medição de cloreto no suor de <30 mmol/L (<30 mEq/L; em todas as faixas etárias) sugere improbabilidade para FC.[29] No entanto, se a dúvida permanecer, recomenda-se o encaminhamento a um centro de FC mesmo na presença de um teste do suor negativo.
Teste de suor positivo: uma medição de cloreto no suor ≥60 mmol/L (≥60 mEq/L) é consistente com FC e requer o encaminhamento imediato a um centro de FC.
Teste do suor intermediário: medição de cloreto no suor de 30-59 mmol/L (30-59 mEq/L); a análise genética do CFTR é necessária.
Os testes do suor podem ser realizados em crianças de qualquer idade. Algumas crianças podem não produzir a quantidade de suor suficiente para fornecer resultados exatos. Se isso ocorrer, a criança deve ser retestada em até uma semana.
Testes genéticos de CFTR
O teste genético pode ajudar a estabelecer o diagnóstico. A maioria dos laboratórios realizará um "rastreamento" inicial para as mutações mais comuns no regulador da condutância transmembrana da fibrose cística (CFTR). Se duas mutações comuns não forem encontradas, a maioria dos laboratórios tem a opção de sequenciar uma porção maior do gene CFTR ou todo ele.
Em indivíduos com resultados do teste do suor que se enquadrarem na faixa intermediária (definida como uma medição de cloreto no suor de 30-59 mmol/L [30-59 mEq/L]), uma avaliação com análise genética do CFTR continua sendo essencial.Subgrupos desses pacientes podem ser caracterizados como portadores da síndrome metabólica relacionada ao CFTR, também conhecida como positivos ao rastreamento e com diagnóstico de FC inconclusivo (CFSPID) na Europa.
Exames fisiológicos e auxiliares
Os testes fisiológicos incluem diferença de potencial nasal e medição da corrente intestinal.Os exames auxiliares incluem exame de imagem dos seios nasais, os quais podem mostrar pansinusite, e swab de orofaringe posterior, que pode demonstrar patógenos respiratórios.[32] Nem o exame de imagem do seio nasal nem o swab de orofaringe são testes específicos para a FC.
Em pacientes com FC que apresentam apendicite, o achado patológico de um material espessado e colorido por hematoxilina dentro das criptas do apêndice é patognomônico.[33]
Rastreamento do neonato
A Cystic Fibrosis Foundation dos EUA recomenda que o rastreamento neonatal adequado seja realizado em todos os bebês.[34] Os painéis de rastreamento de neonatos que incluem a FC não devem substituir o rastreamento pré-gestação ou pré-natal de portadores.[35]
O uso deste conteúdo está sujeito ao nosso aviso legal