Etiologia
Na cetoacidose diabética (CAD), ocorre uma redução na concentração efetiva de insulina circulante, acompanhada por níveis elevados de hormônios contrarreguladores (glucagon, catecolaminas, cortisol e hormônio do crescimento). Essas alterações hormonais são a causa das graves alterações metabólicas características da CAD. Os dois fatores desencadeantes mais comuns são infecção (particularmente infecções do trato urinário e pneumonia) e interrupção ou administração inadequada da insulinoterapia.[1][18] Condições clíncas subjacentes que provocam a liberação de hormônios contrarreguladores, como infarto do miocárdio, AVC ou pancreatite, também podem desencadear a CAD nos indivíduos com diabetes.[18] Além disso, os medicamentos que afetam o metabolismo dos carboidratos, incluindo corticosteroides, diuréticos tiazídicos, simpatomiméticos, antipsicóticos atípicos, inibidores de checkpoint imunológico, cocaína e cannabis, podem contribuir para o desenvolvimento da CAD.[1][19][20][21][22][6][23] O uso de inibidores da proteína cotransportadora de sódio e glicose 2 (SGLT2) e do inibidor duplo SGLT1/SGLT2 sotagliflozina também foi implicado no desenvolvimento de CAD, incluindo a apresentação atípica de cetoacidose euglicêmica, tanto no diabetes do tipo 1 quanto no tipo 2.[1][24][25][26][27][28][29][30] A incidência, no entanto, permanece baixa, com apenas um modesto aumento do risco absoluto. Ensaios clínicos de desfecho cardiovascular com inibidores de SGLT2 relataram taxas de CAD de 0.1% a 0.6%, em comparação com <0.1% a 0.3% nos grupos placebo.[31]
Fisiopatologia
A redução da concentração ou da ação da insulina, juntamente com o aumento dos níveis de hormônios contrarreguladores, leva à hiperglicemia, à depleção de volume e ao desequilíbrio eletrolítico que estão na base da fisiopatologia da cetoacidose diabética.[18] Essas alterações hormonais levam à gliconeogênese hepática e renal, à glicogenólise e à utilização prejudicada da glicose nos tecidos periféricos, resultando em hiperglicemia e hiperosmolaridade. A deficiência de insulina também promove a lipólise, liberando ácidos graxos livres do tecido adiposo, que sofrem oxidação hepática para formar os corpos cetônicos beta-hidroxibutirato e acetoacetato, levando à cetonemia e à acidose metabólica.[18] Estudos demonstraram elevações em citocinas pró-inflamatórias e biomarcadores inflamatórios (por exemplo, proteína C-reativa), bem como marcadores de estresse oxidativo, peroxidação lipídica e risco cardiovascular, durante crises hiperglicêmicas.[32] Essas anormalidades geralmente se normalizam dentro de 24 horas após o início da terapia com insulina e reposição de fluidos.[32] Elevações semelhantes em citocinas pró-inflamatórias e marcadores de estresse oxidativo foram observadas em pessoas sem diabetes durante a hipoglicemia induzida por insulina.[33] Os estados pró-inflamatórios e pró-coagulantes observados tanto em crises hiperglicêmicas quanto em hipoglicemia podem refletir respostas adaptativas ao estresse fisiológico agudo, em vez de efeitos diretos da alteração dos níveis de glicose sanguínea.[33][32]
[Figure caption and citation for the preceding image starts]: Patogênese da cetoacidose diabética (CAD) e estado hiperosmolar hiperglicêmico (EHH); os fatores desencadeantes incluem estresse, infecção e insuficiência de insulina. AGL: ácido graxo livre; SHH: estado hiperosmolar hiperglicêmicoDe: Kitabchi AE, Umpierrez GE, Miles JM, et al. Diabetes Care. 2009,32:1335-43; usado com permissão [Citation ends].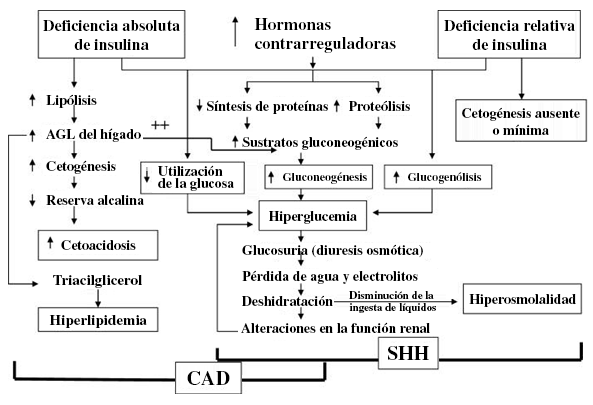
O uso deste conteúdo está sujeito ao nosso aviso legal