Etiologia
As cardiomiopatias podem ser subdivididas de acordo com a classificação desenvolvida pelo grupo de trabalho da American Heart Association.[1]
Cardiomiopatias primárias: genéticas
Cardiomiopatia hipertrófica
Uma condição relativamente comum que afeta cerca de 1 em 500 da população em geral, entre os grupos raciais.[5][6] A cardiomiopatia hipertrófica (CMH) é caracterizada pelo desenvolvimento de um ventrículo esquerdo (VE) não dilatado hipertrofiado, na ausência de outro fator predisponente (como estenose aórtica ou hipertensão).[Figure caption and citation for the preceding image starts]: Cardiomiopatia hipertrófica apical: vista ecocardiográfica de 4 câmaras com contrasteAhmed I, Smalley SJ, Zhu DWX, et al. Sudden cardiac arrest in apical hypertrophic cardiomyopathy. BMJ Case Reports. 2009;doi:10.1136/bcr.04.2009.17 [Citation ends].
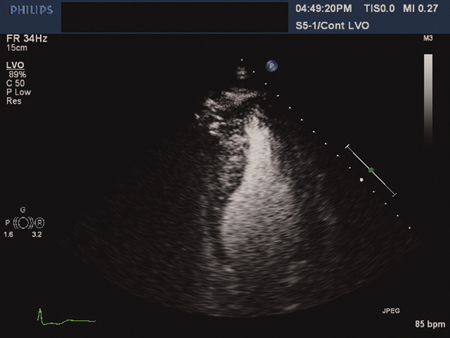 O desenvolvimento da hipertrofia está caracteristicamente relacionado à idade. Embora trabalhos iniciais tenham sugerido que os indivíduos geralmente desenvolvem hipertrofia durante a adolescência, é cada vez maior o reconhecimento sobre o desenvolvimento da doença com início tardio. Com isso, os membros adultos e adolescentes da família não afetados clinicamente devem receber orientação para realizar o rastreamento de acordo com as diretrizes atuais.[7][8][9] As diretrizes da European Society of Cardiology (ESC) oferecem aos profissionais da saúde um esquema prático de diagnóstico e tratamento para pacientes de todas as idades.[7][8] As diretrizes também consideram as implicações do diagnóstico para as famílias.
O desenvolvimento da hipertrofia está caracteristicamente relacionado à idade. Embora trabalhos iniciais tenham sugerido que os indivíduos geralmente desenvolvem hipertrofia durante a adolescência, é cada vez maior o reconhecimento sobre o desenvolvimento da doença com início tardio. Com isso, os membros adultos e adolescentes da família não afetados clinicamente devem receber orientação para realizar o rastreamento de acordo com as diretrizes atuais.[7][8][9] As diretrizes da European Society of Cardiology (ESC) oferecem aos profissionais da saúde um esquema prático de diagnóstico e tratamento para pacientes de todas as idades.[7][8] As diretrizes também consideram as implicações do diagnóstico para as famílias.A hipertrofia geralmente é assimétrica, mas é importante reconhecer que ela pode acometer qualquer parte do miocárdio. É frequente a disfunção diastólica. Até um terço dos pacientes apresenta obstrução de saída do ventrículo esquerdo em repouso, enquanto outros podem ter alterações hemodinâmicas induzidas por exercícios físicos ou torção.
Arritmias atriais são comuns, e os indivíduos acometidos apresentam risco elevado de tromboembolismo.[10]
Aproximadamente 8% dos indivíduos desenvolverão disfunção sistólica do ventrículo esquerdo.[11]
O diagnóstico geralmente é feito mediante ecocardiografia (ECG) combinada a exames de imagem cardíaca, mais comumente a ecocardiografia inicialmente.[8][12][13][14] Investigações adicionais (monitoramento ambulatorial, teste ergométrico) podem ser apropriadas para a estratificação de risco dos pacientes com CMH.[15] Essas investigações são particularmente relevantes em indivíduos com qualquer um dos seguintes fatores de risco: parada cardíaca prévia ou taquicardia ventricular sustentada, taquicardia ventricular não sustentada, hipertrofia ventricular esquerda extrema (>30 mm), síncope inexplicada, síncope inexplicável, resposta anormal da pressão arterial ao exercício, história familiar de morte súbita e obstrução significativa da via de saída do ventrículo esquerdo.[16][17][18][19]
A ressonância nuclear magnética (RNM) cardíaca é cada vez mais usada, pois proporciona uma estimativa acurada sobre a extensão da hipertrofia, e a fibrose miocárdica pode ser detectada como um realce tardio com gadolínio (RTG).[13][20] Estudos mostram que a extensão do RTG sugere um maior risco de morte súbita cardíaca, mas não constitui um preditor independente. O RTG está associado a uma maior incidência de taquicardia ventricular não sustentada.[21] Investigadores dos EUA observaram que a presença do gadolínio tardio pode permitir uma melhor seleção dos indivíduos que precisam de desfibriladores para prevenção primária.[22] As diretrizes da ESC recomendam avaliar os pacientes com CMH por meio de uma RNM cardíaca e que a presença e a extensão do RTG podem ser consideradas ao se aconselhar os pacientes sobre o risco de morte súbita.[8] As diretrizes dos EUA recomendam a RNM cardíaca como uma investigação complementar após a ecocardiografia, ou como uma alternativa à ecocardiografia para aqueles pacientes nos quais a ecocardiografia for inconclusiva.[9]
Um modelo de predição de risco da ESC, conhecido como HCM Risk-SCD, foi desenvolvido e validado para oferecer uma estimativa individualizada do risco de morte súbita cardíaca em 5 anos.[23] ESC: HCM Risk-SCD Opens in new window
Pode ser necessária uma angiografia coronária para descartar doença arterial coronariana concomitante em pacientes com mais de 40 anos de idade ou que tenham outros fatores de risco coronários, em particular quando apresentam sintomas típicos de angina ou dispneia. Embora o desarranjo das fibras miocárdicas seja uma característica patológica da CMH, raramente a biópsia endomiocárdica é usada, pois o desarranjo das fibras miocárdicas muitas vezes é irregular, e uma pequena quantidade de desarranjo pode ser encontrada em indivíduos normais.
A função do teste genético ultrapassa o âmbito deste tópico. No entanto, a CMH é descrita tradicionalmente como uma doença sarcomérica com mutações relatadas nos genes que codificam muitas proteínas sarcoméricas, incluindo: cadeia pesada da beta-miosina, proteína C de ligação de miosina, troponina T, troponina I e actina. Genes não sarcoméricos incluem o PRKAG2 (frequentemente associado à pré-excitação) e raramente o LAMP-2 (doença de Danon).[24] É importante reconhecer que muitas outras condições podem apresentar um fenótipo de CMH. Dentre essas condições estão algumas sindrômicas, como a doença de Fabry (particularmente em homens acima de 35 anos de idade com hipertrofia concêntrica), síndromes de Noonan/Leopard e ataxia de Friedreich, bem como vários outros distúrbios metabólicos, doenças mitocondriais e doenças de depósito de glicogênio.[25][26][27] O diagnóstico dessas condições requer um alto índice de suspeita clínica e o uso de ferramentas diagnósticas apropriadas.[28] Um importante diagnóstico diferencial em indivíduos jovens saudáveis é o "coração de atleta". Vários fatores podem ser usados para distinguir a CMH do coração de atleta, como: extensão da hipertrofia, tamanho da cavidade do VE, extensão do aumento do átrio esquerdo, presença de obstrução na via de saída do VE, alterações em parâmetros derivados do Doppler tecidual, pico de consumo do volume de oxigênio (VO2), níveis de pró-peptídeo natriurético do tipo B N-terminal (NT-proPNB), regressão na cessação de exercícios e teste genético.[29]
Cardiomiopatia arritmogênica do ventrículo direito
Anteriormente conhecida como displasia/cardiomiopatia arritmogênica do ventrículo direito.[30] Caracterizada pela substituição progressiva do miocárdio por material fibroadiposo. Essas alterações são encontradas mais comumente no triângulo de displasia (entrada, saída e ápice do ventrículo direito), mas são cada vez mais reconhecidas no VE. Elas incluem variantes clínicas nas quais o grau de envolvimento do ventrículo esquerdo é igual ou superior à gravidade do envolvimento do ventrículo direito, denominada cardiomiopatia arritmogênica biventricular ou dominante esquerda, respectivamente.[31]
A cardiomiopatia arritmogênica do ventrículo direito representa um amplo espectro da doença, envolvendo anormalidades eletrofisiológicas e funcionais. As manifestações eletrofisiológicas variam desde anormalidades eletrocardiográficas mais modestas até arritmias ventriculares de risco de vida. Similarmente, anormalidades da função muscular variam desde alterações contráteis menores até insuficiência ventricular esquerda e direita grave.[32] Os pacientes apresentam uma ampla variedade de sintomas, desde arritmias até insuficiência cardíaca e morte súbita. Na região do Vêneto, na Itália, a cardiomiopatia arritmogênica do ventrículo direito é a causa mais comum de morte súbita cardíaca em indivíduos jovens.[33]
O diagnóstico de cardiomiopatia arritmogênica do ventrículo direito é difícil e requer várias investigações, incluindo: exame de imagem com ecocardiografia/RNM/angiografia, monitoramento ambulatorial e teste ergométrico para verificar arritmias, presença de ECG anormal em repouso (anormalidades na onda T, derivações tardias de condução do ramo direito, ondas épsilon), potencial tardio em um ECG de alta resolução e alterações histopatológicas observadas em uma biópsia endomiocárdica ou post mortem.
A herança autossômica dominante é o padrão mais comum, embora variantes autossômicas recessivas (geralmente com alterações de pele e cabelo, como na doença de Naxos e na síndrome de Carvajal) também sejam observadas. Vários genes foram implicados, mais comumente os genes desmossomais, embora também haja relatos de mutações nos genes RYR2 e do fator de transformação de crescimento β3 (TGF-β3).[34]
Os critérios Task Force para o diagnóstico de cardiomiopatia arritmogênica do ventrículo direito/displasia foram publicados em 1994.[35] Entretanto, esses critérios foram considerados muito restritivos ao lidar com outros membros da família, por isso, foram propostos critérios modificados para esses indivíduos.[36] Mais recentemente, foram propostas outras modificações nesses critérios, incluindo a avaliação da RNM da estrutura cardíaca e o fornecimento de medições quantificáveis adicionais do tamanho e da função do ventrículo direito (VD) associados à cardiomiopatia arritmogênica do ventrículo direito.[30] Há informações limitadas sobre a história natural da doença, embora quatro estágios sejam geralmente reconhecidos.[37]
Oculto: fase inicial, geralmente assintomática, embora possa manifestar-se como morte súbita.
Distúrbios elétricos evidentes: fase instável com arritmias sintomáticas, geralmente bloqueio de ramo esquerdo, com origem sugestiva no VD.
Insuficiência no VD: fase de deterioração progressiva da função contrátil do VD.
Falha na bomba biventricular: fase de dilatação biventricular progressiva, a qual pode mimetizar aquela observada na cardiomiopatia dilatada (CMD) idiopática.
Canalopatias iônicas
Incluem síndrome do QT longo (herança autossômica dominante ou recessiva), síndrome de Brugada, taquicardia ventricular polimórfica catecolaminérgica (forma autossômica dominante ou recessiva), síndrome do QT curto (herança autossômica dominante), fibrilação ventricular idiopática e doença do nó sinusal (herança autossômica dominante). É importante notar que o grupo de trabalho da European Society of Cardiology não concorda com a inclusão essas canalopatias como parte do espectro das cardiomiopatias.[3]
Outras cardiomiopatias genéticas primárias incluem doença do sistema de condução e miopatias mitocondriais.
Cardiomiopatias primárias: mistas
Cardiomiopatia dilatada[Figure caption and citation for the preceding image starts]: Cardiomiopatia dilatada: ecocardiografiaTanejal AK, Wong J, Bayliss J. Antipsychotic-drug-induced dilated cardiomyopathy. BMJ Case Reports. 2009; doi:10.1136/bcr.09.2008.0958 [Citation ends].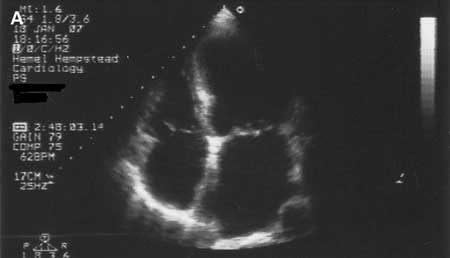
Caracterizada pela presença de dilatação e disfunção sistólica do ventrículo esquerdo na ausência de condições anormais de carga ou doença arterial coronariana significativa. A dilatação do ventrículo direito também é frequente. A prevalência de cardiomiopatia dilatada é de aproximadamente 1 em 250.[5][6] Estima-se que 30% a 40% dos casos sejam familiares; relatam-se padrões de heranças autossômica dominante, autossômica recessiva, ligada ao cromossomo X e mitocondrial.[6] Também deve haver a suspeita de uma doença familiar quando houver história familiar de morte súbita, doença do sistema de condução ou miopatia esquelética associada. O teste genético desses pacientes é recomendado porque certas mutações (por exemplo, no gene LMNA) podem indicar um risco maior.[5]
A European Society of Cardiology (ESC) propôs uma definição revisada que separa a cardiomiopatia dilatada em duas formas de acordo com os parâmetros de imagem: cardiomiopatia dilatada e cardiomiopatia não dilatada do ventrículo esquerdo.[8][38]
A cardiomiopatia não dilatada do ventrículo esquerdo é caracterizada por cicatrização não isquêmica ou substituição gordurosa do VE na ausência de dilatação e pode ocorrer com ou sem anormalidades globais ou regionais de contratilidade da parede ou hipocinesia isolada do VE sem cicatrização que não seja explicada por condições de carga anormais ou doença arterial coronariana.[8][38]
Recomenda-se a avaliação dos pacientes com suspeita de cardiomiopatia não dilatada do ventrículo esquerdo com ECG, ecocardiografia e RNM cardíaca.
Há uma sobreposição significativa nas variantes genéticas implicadas na cardiomiopatia não dilatada do ventrículo esquerdo com aquelas conhecidas por causar CMD e cardiomiopatia arritmogênica do ventrículo direito.
Como essa categoria de cardiomiopatia é relativamente nova, pouco se sabe sobre a prevalência e os desfechos dessa afecção. Acredita-se que o genótipo influencie fortemente o risco de morte súbita com as variantes genéticas de alto risco, incluindo: DSP, FLNC, LMNA, DES e RBM20.[8]
Vários dos genes sarcoméricos implicados no desenvolvimento de cardiomiopatia hipertrófica também podem causar um fenótipo de cardiomiopatia dilatada.[39] Mutações no gene lamina A/C podem resultar em diferentes fenótipos. Entretanto, da perspectiva cardíaca, o diagnóstico de laminopatia é importante, pois indivíduos acometidos apresentam risco elevado de doença do sistema da condução progressiva e morte súbita.[40] Uma extensa lista de condições que podem causar cardiomiopatia dilatada foi reconhecida. Uma causa bem conhecida é a pós-miocardite, que frequentemente é assintomática da perspectiva dos pacientes. As outras causas incluem: bebidas alcoólicas; agentes quimioterápicos (antraciclinas, trastuzumabe); doenças de depósito (por exemplo, hemocromatose); distúrbios autoimunes e sistêmicos; distúrbios neuromusculares; distúrbios mitocondriais, metabólicos/endócrinos (tireoide, diabetes, acromegalia, feocromocitoma) e distúrbios nutricionais (em particular, problemas de desnutrição por tiamina, selênio e proteínas).[41] Se nenhuma causa for identificada apesar de uma investigação extensa, o termo cardiomiopatia dilatada idiopática é aplicado.
Cardiomiopatia isquêmica
Uma consequência comum da isquemia miocárdica que causa disfunção cardíaca. É importante observar que isso está excluído da classificação descrita na declaração cientifica da American Heart Association.[1]
Cardiomiopatia restritiva
Trata-se de uma cardiomiopatia menos bem definida, pois seu diagnóstico baseia-se na presença de um padrão de enchimento ventricular restritivo. O grupo de trabalho da European Society of Cardiology a define como: "fisiologia restritiva na presença de volumes diastólicos normais ou reduzidos, volumes sistólicos normais ou reduzidos e espessura normal da parede".[3] De muitas formas, trata-se de uma descrição do estado da fisiopatologia, que pode representar várias patologias ou cardiomiopatias, em vez de uma cardiomiopatia distinta.
Pode ser idiopática, familiar (foi relacionada a mutações no gene da troponina I ou da desmina; no caso da desmina, frequentemente em associação com uma miopatia esquelética) ou associada a vários distúrbios sistêmicos, como hemocromatose, amiloidose, sarcoidose, doença de Fabry, síndrome carcinoide, esclerodermia, toxicidade da antraciclina ou radiação prévia. Na população pediátrica, algumas síndromes metabólicas raras (doença de Gaucher ou síndrome de Hurler) devem ser descartadas.
A patologia endocardial (síndromes hipereosinofílicas ou fibrose endomiocardial) também pode resultar em uma cardiomiopatia restritiva.[42] Os pacientes podem apresentar sintomas de dispneia ou palpitações, e esses sintomas estão relacionados à disfunção diastólica. Geralmente, a pressão venosa jugular é elevada. O eletrocardiograma (ECG) geralmente é normal, com evidência de hipertrofia biatrial e alterações inespecíficas nas ondas ST-T. Imagens cardíacas por ecocardiografia ou RNM mostram aumento biatrial com evidência de disfunção diastólica, evidente em padrões de dopplerfluxometria mitral, bem como sinais derivados do Doppler tecidual.
Como pode apresentar-se de maneira similar à pericardite constritiva, a cardiomiopatia restritiva deve ser diferenciada dessa doença. Para isso, pode ser necessário o cateterismo cardíaco. A biópsia endomiocárdica pode ser útil em casos selecionados.[43] São usados diuréticos para o controle sintomático, mas com cuidado, pois eles podem reduzir a pré-carga e atuar de maneira contraprodutiva. Os pacientes podem ter arritmias complexas, inclusive fibrilação atrial, e cardioversores-desfibriladores implantáveis (CDIs) podem ser necessários para o manejo de taquiarritmias ventriculares.
Cardiomiopatias primárias: adquiridas
Miocardite inflamatória
Doença inflamatória adquirida, aguda ou crônica, que acomete o miocárdio e que pode ser causada por vários fatores, incluindo agentes infecciosos, toxinas e medicamentos. Três fases são reconhecidas: ativa, em processo de cura e curada. Os critérios patológicos padrão de Dallas requerem que um infiltrado inflamatório, com ou sem necrose miocítica, esteja presente na avaliação histológica padrão.[44] Entretanto, critérios mais recentes atribuem os estágios da miocardite com base nos fatores de risco, sintomas, marcadores de imagem e histopatológicos, além das instabilidades hemodinâmica e elétrica.[45]
Há riscos significativos associados à biópsia endomiocárdica, e isso tem acarretado recomendações com base na probabilidade de encontrar doenças específicas.[46] Dois cenários que descrevem as apresentações mais comuns de miocardite fulminante e miocardite de células gigantes são: 1) insuficiência cardíaca inicial inexplicada com <2 semanas de duração, combinada com um ventrículo esquerdo dilatado ou de tamanho normal e comprometimento hemodinâmico; e 2) insuficiência cardíaca inicial inexplicada de 2 semanas a 3 meses de duração, associada a um ventrículo esquerdo dilatado e evidência de arritmias ventriculares ou bloqueio atrioventricular de alto grau ou falha em responder aos cuidados habituais dentro de 1 a 2 semanas. A RNM cardíaca pode fornecer uma ferramenta diagnóstica alternativa, pois os estudos mostram uma boa correlação entre regiões ativas de miocardite e áreas de intensidade de sinal anormal na RNM.[13]
Síndrome do takotsubo
Doença manifestada por sintomas e sinais de infarto agudo do miocárdio na ausência de doença arterial coronariana significativa ou espasmo. (O nome da doença faz referência a um vaso-armadilha comumente utilizado por pescadores no Japão para a captura de polvos, de gargalo estreito e fundo alargado, semelhante ao aspecto visual do coração.)[47] Alguns autores referem-se à condição como "balonamento apical transitório e cardiomiopatia de estresse".[Figure caption and citation for the preceding image starts]: Ventriculograma esquerdo demonstrando balonamento apical na cardiomiopatia de TakotsuboAugustine DX, Domanski A, Garg A. The stress of chest pain: a case of tako-tsubo cardiomyopathy. BMJ Case Reports. 2009; doi:10.1136/bcr.03.2009.1660 [Citation ends].
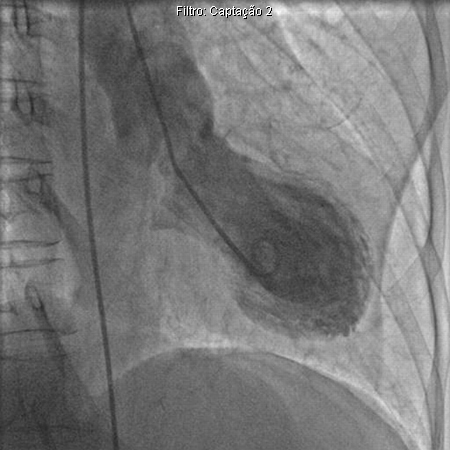
Os pacientes apresentam dor torácica associada a supradesnivelamento do segmento ST, inversão da onda T e elevações leves em marcadores cardíacos. Essa dor é mais comumente observada em mulheres menopausadas e geralmente é precedida por alguma forma de estresse emocional ou físico significativo.[48] Durante o evento agudo, elevações transitórias em níveis de noradrenalina são registrados (na ausência de feocromocitoma). Se o paciente sobreviver ao evento agudo, a função ventricular esquerda geralmente será normalizada dentro de várias semanas.
Vários grupos propuseram critérios de diagnóstico. Os critérios usados mais amplamente são aqueles de um grupo da Mayo Clinic (critérios de diagnóstico inicialmente relatados em 2004 e modificados em 2008):[47]
Hipocinesia, acinesia ou discinesia transitória de segmentos intermediários do ventrículo esquerdo com ou sem envolvimento apical; anormalidades regionais da parede estendem-se além de uma única distribuição vascular epicárdica; é comum haver fatores desencadeantes estressantes, mas isso nem sempre ocorre
Ausência de doença coronariana obstrutiva ou evidência angiográfica de ruptura aguda de placa
Novas anormalidades eletrocardiográficas (supradesnivelamento do segmento ST e/ou inversão da onda T) ou elevação modesta na troponina cardíaca
Ausência de feocromocitoma/miocardite.
Periparto
A cardiomiopatia periparto (CMPP), definida pelo grupo de trabalho sobre a CMPP da European Society of Cardiology, é uma cardiomiopatia idiopática geralmente apresentando insuficiência cardíaca secundária à disfunção sistólica ventricular esquerda (FEVE <45%), no fim da gravidez ou nos meses após o parto, quando nenhuma outra causa de insuficiência cardíaca é encontrada.[49][50] É incomum, ocorrendo em aproximadamente 1 em 4000 nascimentos.[51] Relatos sugerem que é mais comum em mulheres com mais de 30 anos e existe associação com hipertensão gestacional e gravidez gemelar. A CPPM parece ser muito mais comum em mulheres de descendência afro-americana, com baixa incidência entre mulheres hispânicas. Algumas pacientes apresentam a doença no início da gestação. Essas pacientes foram classificadas como portadoras de cardiomiopatia associada ao início da gestação.[52]
Vários mecanismos (como miocardite, causas imunológicas e hemodinâmicas) foram propostos. A doença pode estar associada à recuperação completa em até 50% dos casos; em outros, pode evoluir para insuficiência cardíaca grave, resultando em transplante ou morte cardíaca. Parece ser responsável por morbidade significativa.[53] O estresse oxidativo e a geração de um subfragmento cardiotóxico de prolactina podem desempenhar papéis fundamentais na fisiopatologia da CMPP. Dessa forma, o bloqueio farmacológico da prolactina oferece a possibilidade de uma terapia específica para a doença.[49]
Induzida por taquicardia
Em determinados indivíduos, períodos prolongados de frequência cardíaca rápida associados à taquicardia atrial, fibrilação atrial ou arritmias ventriculares podem causar cardiomiopatia relacionada à taquicardia.[54] Trata-se de uma importante condição a ser reconhecida, pois a cardiomiopatia pode ser reversível quando o controle de frequência cardíaca é obtido.[55]
Causas secundárias de cardiomiopatia
As cardiomiopatias secundárias demonstram comprometimento miocárdico no contexto de distúrbios sistêmicos (multiorgânicos) generalizados.[1]
Doenças infiltrantes ou de depósito geralmente resultam em cardiomiopatia. As causas são descritas a seguir.
Amiloidose (hipertrófica ou restritiva): caracterizada pelo depósito de fibrilas amiloides no coração. Inclui uma ampla gama de doenças, como amiloidose primária, polineuropatia amiloide familiar e amiloidose cardíaca senil.[56] Os pacientes apresentam sintomas e sinais característicos de insuficiência cardíaca ou distúrbios do ritmo cardíaco.[57][58] O diagnóstico pode ser suspeitado quando se realiza um ECG ou uma ecocardiografia como parte da investigação cardíaca inicial do paciente. O ECG se caracteriza pela presença de voltagens pequenas no QRS, padrão de pseudoinfarto nas derivações anteriores ou inferiores e arritmias ventriculares e supraventriculares. A ecocardiografia é notável por um maior espessamento do septo atrial e da parede do ventrículo esquerdo; aumento da ecogenicidade miocárdica (pontilhada), associada a algum espessamento valvar; e frequentemente um pequeno derrame pericárdico. Um dos traços característicos da doença é a relação inversa entre a massa do VE na imagem cardíaca e a voltagem no ECG.[57][59] A RNM cardíaca é cada vez mais realizada como ferramenta diagnóstica.[8][13][57][60] A RNM revela uma diminuição difusa do sinal em T1 e T2 do miocárdio, correspondendo a infiltração amiloide, e um aspecto característico do RTG subendocárdico global. A biópsia de um tecido acometido geralmente fornece confirmação histológica.[57][59]
Doença de Gaucher (restritiva).
Mucopolissacaridose (síndrome de Hunter e doença de Hurler; principalmente cardiomiopatia dilatada).[61]
Hemocromatose (cardiomiopatia dilatada ou restritiva): a hemocromatose clássica é uma doença autossômica recessiva associada a uma mutação no gene HFE, que causa sobrecarga de ferro. Trata-se de um distúrbio multissistêmico, no qual o envolvimento cardíaco é manifesto pelo desenvolvimento de uma cardiomiopatia dilatada, causando insuficiência cardíaca e distúrbio do sistema de condução.[58] O tratamento com depleção de ferro pode reverter alterações precoces. O envolvimento cardíaco e a detecção de ferro são facilmente constatados pela RNM cardíaca.[8][13]
Doença de Fabry (cardiomiopatia restritiva ou hipertrófica).
Doença de Pompe (doença de depósito de glicogênio tipo 2; cardiomiopatia hipertrófica).
Doença de Niemann-Pick (cardiomiopatia hipertrófica).
Estados de sobrecarga férrica, incluindo talassemia.
Várias toxinas estão associadas ao desenvolvimento de cardiomiopatia.
Há forte correlação entre a doxorrubicina e o desenvolvimento de cardiomiopatia.[62] A cardiomiopatia também pode resultar de terapia com trastuzumabe.[63]
O consumo crônico de bebidas alcoólicas pode danificar diretamente o tecido miocárdico, induzindo à apoptose ou pode estar associado à deficiência de tiamina e ao desenvolvimento de beribéri úmido (embora o mecanismo exato seja desconhecido).[64]
O cobalto adicionado à cerveja e a exposição a determinadas substâncias químicas, como os metais pesados, também podem causar danos ao miocárdio.[65]
As endocrinopatias são conhecidas por sua associação à cardiomiopatia, embora as evidências sejam limitadas a relatos de caso, e a cardiomiopatia resultante pode ser dilatada ou restritiva.
O diabetes mellitus pode causar cardiomiopatia infiltrante/restritiva em razão da glicosilação do miocárdio e da subsequente disfunção diastólica.[66] É importante descartar doença coronariana associada em pacientes com diabetes mellitus que apresentam suspeita de cardiomiopatia.
A cardiomiopatia também pode ser causada por hipertireoidismo/hipotireoidismo, hiperparatireoidismo, feocromocitoma e acromegalia.
As cardiomiopatias constituem uma característica reconhecida de determinadas síndromes cardiofaciais.
Síndrome de Noonan.
Lentiginose.[26]
Deficiências nutricionais de várias vitaminas e minerais são associadas ao desenvolvimento de cardiomiopatia.
A deficiência de tiamina causa insuficiência cardíaca de alto débito, conhecida como beribéri úmido. Muitos mecanismos fisiopatológicos foram propostos, mas considera-se que a lesão mitocondrial inicial possa causar, subsequentemente, cardiomiopatia.[67]
Outras deficiências nutricionais envolvendo vitamina C, niacina, selênio e vitamina D também podem causar cardiomiopatia.
Outras causas
Distúrbios neuromusculares e neurológicos, como ataxia de Friedreich, distrofias musculares (distrofia muscular de Duchenne, Becker e Emery-Dreifuss; distrofia miotônica) podem apresentar envolvimento cardíaco.
Distúrbios autoimunes, como lúpus eritematoso sistêmico.
A sarcoidose é um distúrbio granulomatoso sistêmico de causa desconhecida. O envolvimento cardíaco manifesto é incomum, e os pacientes podem apresentar insuficiência cardíaca (em razão do desenvolvimento de cardiomiopatia), bradiarritmias/bloqueio atrioventricular de alto grau ou taquiarritmias ventriculares, os quais causam morte súbita.[68] O envolvimento miocárdico subclínico é comum, e o envolvimento cardíaco é frequentemente apenas identificado na autópsia. O envolvimento granulomatoso geralmente é em placas, e o rendimento da biópsia endomiocárdica é relativamente baixo. Vários critérios de diagnóstico foram desenvolvidos.[69] Os pacientes com sarcoidose cardíaca e sintomas de síncope devem ser submetidos a uma investigação profunda e ser considerados para terapia com dispositivos, incluindo CDI. A RNM cardíaca está sendo cada vez mais usada como uma técnica de imagem útil e a FDG-PET pode ser útil tanto para a identificação de sarcoidose cardíaca quanto para monitorar a atividade da doença.[13][70][71][72][73] Podem ser necessários encaminhamentos a outros especialistas para um tratamento imunossupressor da doença subjacente.
Desequilíbrio eletrolítico: geralmente potássio, fosfato ou magnésio.
Fibrose endomiocárdica.
Endocardite de Loeffler/síndrome hipereosinofílica.
O uso deste conteúdo está sujeito ao nosso aviso legal