Doenças do armazenamento lisossomal hereditárias comuns
- Visão geral
- Teoria
- Diagnóstico
- Tratamento
- ACOMPANHAMENTO
- Recursos
Algoritmo de tratamento
Observe que as formulações/vias e doses podem diferir entre nomes e marcas de medicamentos, formulários de medicamentos ou localidades. As recomendações de tratamento são específicas para os grupos de pacientes:ver aviso legal
Doença de Gaucher do tipo 1
terapia de suporte
Uma abordagem multidisciplinar é essencial, pois essas doenças são distúrbios multissistêmicos; muitos especialistas diferentes podem estar envolvidos no cuidado de pacientes individuais. É recomendável o envolvimento precoce de centros especializados; os cuidados devem ser coordenados pelo centro.
O médico deve monitorar doença esquelética, neoplasias hematológicas, anormalidades metabólicas e endócrinas, doença de Parkinson e cirrose hepática, além de controlar essas complicações à medida que surgem.
terapia de reposição enzimática
Tratamento adicional recomendado para ALGUNS pacientes no grupo de pacientes selecionado
Os pacientes devem ser tratados sob a supervisão de especialistas. A redução da dose pode ser possível após os pacientes se estabilizarem.
Pacientes assintomáticos não exigem tratamento.[99]Kaplan P, Baris H, De Meirleir L, et al. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr. 2013 Apr;172(4):447-58. http://www.ncbi.nlm.nih.gov/pubmed/22772880?tool=bestpractice.com No entanto, a terapia de reposição enzimática (TRE) deve ser considerada em todas as crianças sintomáticas, e em adultos com reduções significativas nas contagens sanguíneas (por exemplo, nível de hemoglobina <100 g/L [10 g/dL], plaquetas <100 x 10⁹/L), aumento significativo de órgãos (por exemplo, tamanho do baço >10x o normal), presença de doença esquelética demonstrada na ressonância nuclear magnética e/ou qualquer outro dano a órgãos (por exemplo, evidência de lesão do pulmão).[99]Kaplan P, Baris H, De Meirleir L, et al. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr. 2013 Apr;172(4):447-58. http://www.ncbi.nlm.nih.gov/pubmed/22772880?tool=bestpractice.com [100]Hughes D, Cappellini MD, Berger M, et al. Recommendations for the management of the haematological and onco-haematological aspects of Gaucher disease. Br J Haematol. 2007 Sep;138(6):676-86. https://onlinelibrary.wiley.com/doi/10.1111/j.1365-2141.2007.06701.x http://www.ncbi.nlm.nih.gov/pubmed/17655728?tool=bestpractice.com A TRE tem benefício comprovado na melhora de anormalidades hematológicas e dores ósseas, e na redução do tamanho do fígado e baço. Densidade óssea, função pulmonar e qualidade de vida também melhoram.[106]Barton NW, Brady RO, Dambrosia JM, et al. Replacement therapy for inherited enzyme deficiency - macrophage-targeted glucocerebrosidase for Gaucher's disease. N Engl J Med. 1991 May 23;324(21):1464-70. http://www.ncbi.nlm.nih.gov/pubmed/2023606?tool=bestpractice.com [107]Weinreb NJ, Charrow J, Andersson HC, et al. Effectiveness of enzyme replacement therapy in 1028 patients with type I Gaucher disease after 2 to 5 years of treatment: a report from the Gaucher Registry. Am J Med. 2002 Aug 1;113(2):112-9. http://www.ncbi.nlm.nih.gov/pubmed/12133749?tool=bestpractice.com [108]Gabrowski GA, Kolodny EH, Weinreb NJ, et al. Gaucher disease: phenotypic and genetic variation. In: Scriver CR, Beaudet AL, Sly WS, et al., eds. The metabolic and molecular basis of inherited disease. 9th ed. New York, NY: McGraw-Hill; 2006.
A taliglucerase é uma forma de TRE derivada de plantas. Em 2012 a Food and Drug Administration dos EUA aprovou seu uso para a doença de Gaucher, mas o Comitê de Medicamentos de Uso Humano (CHMP) da European Medicines Agency (EMA) recomendou contra a autorização para comercialização.
A TRE foi associada a reações de hipersensibilidade graves, anafilaxia e reações relacionadas com à infusão. Considere a pré-medicação com um anti-histamínico, antipirético e/ou corticosteroide.
Opções primárias
imiglucerase: crianças e adultos: 60 unidades/kg por via intravenosa a cada 2 semanas inicialmente, ajustar de acordo com a resposta
ou
velaglucerase alfa: crianças com ≥4 anos de idade e adultos: 60 unidades/kg por via intravenosa a cada 2 semanas inicialmente, ajustar de acordo com a resposta
ou
taliglucerase alfa: crianças com ≥4 anos de idade e adultos: 60 unidades/kg por via intravenosa a cada 2 semanas inicialmente, ajustar de acordo com a resposta
terapia de redução de substrato
Tratamento adicional recomendado para ALGUNS pacientes no grupo de pacientes selecionado
É adequada para pacientes afetados menos gravemente que não conseguem tolerar a terapia intravenosa ou que são alérgicos às preparações da terapia de reposição enzimática disponíveis.
Mostrou-se que a terapia de redução de substrato melhora a anemia, a trombocitopenia, o aumento do fígado/baço e a osteoporose.[109]Mistry PK, Lukina E, Ben Turkia H, et al. Effect of oral eliglustat on splenomegaly in patients with Gaucher disease type 1: the ENGAGE randomized clinical trial. JAMA. 2015 Feb 17;313(7):695-706. http://jama.jamanetwork.com/article.aspx?articleid=2110969 http://www.ncbi.nlm.nih.gov/pubmed/25688781?tool=bestpractice.com [110]Cox TM, Aerts JM, Andria G, et al. The role of the iminosugar N-butyldeoxynorjirimycin (miglustat) in the management of type 1 (non-neuronopathic) Gaucher disease: a position statement. J Inherit Metab Dis. 2003;26(6):513-26. http://www.ncbi.nlm.nih.gov/pubmed/14605497?tool=bestpractice.com [111]Cox T, Lachmann R, Hollak C, et al. Novel oral treatment of Gaucher's disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet. 2000 Apr 29;355(9214):1481-5. http://www.ncbi.nlm.nih.gov/pubmed/10801168?tool=bestpractice.com [112]Pastores GM, Giraldo P, Cherin P, et al. Goal-oriented therapy with miglustat in Gaucher disease. Curr Med Res Opin. 2009 Jan;25(1):23-37. http://www.ncbi.nlm.nih.gov/pubmed/19210136?tool=bestpractice.com [113]Poole RM. Eliglustat: first global approval. Drugs. 2014 Oct;74(15):1829-36. http://www.ncbi.nlm.nih.gov/pubmed/25239269?tool=bestpractice.com
Opções primárias
miglustate: adultos: 100 mg por via oral três vezes ao dia, ajustar de acordo com a resposta
Mais miglustateExistem várias marcas de miglustate disponíveis; certifique-se de usar a marca correta para essa indicação.
ou
eliglustate: adultos: a dose depende da genotipagem do CYP2D6 e da administração conjunta de medicamentos que interagem; consulte um especialista para obter orientação quanto à dose
Mais eliglustateEstabeleça o genótipo do paciente usando um exame validado para determinar a genotipagem do CYP2D6 antes de iniciar a terapia.[169]National Institute for Health and Care Excellence. Eliglustat for treating type 1 Gaucher disease. Jun 2017 [internet publication]. https://www.nice.org.uk/guidance/hst5 O eliglustate pode estar aprovado para crianças ≥6 anos de idade em alguns países fora dos EUA.
doença de Gaucher do tipo 2
terapia de suporte
Uma abordagem multidisciplinar é essencial, pois essas doenças são distúrbios multissistêmicos; muitos especialistas diferentes podem estar envolvidos no cuidado de pacientes individuais. É recomendável o envolvimento precoce de centros especializados; os cuidados devem ser coordenados pelo centro.
A doença de Gaucher do tipo 2 é essencialmente intratável. O médico deve monitorar e tratar convulsões, atraso do neurodesenvolvimento e distúrbios da motilidade ocular.
A terapia de reposição enzimática não é efetiva.
doença de Gaucher do tipo 3
terapia de suporte
Uma abordagem multidisciplinar é essencial, pois essas doenças são distúrbios multissistêmicos; muitos especialistas diferentes podem estar envolvidos no cuidado de pacientes individuais. É recomendável o envolvimento precoce de centros especializados; os cuidados devem ser coordenados pelo centro.
O médico deve monitorar e tratar complicações de distúrbios da motilidade ocular, atraso do neurodesenvolvimento e doença esquelética.
terapia de reposição enzimática
Tratamento adicional recomendado para ALGUNS pacientes no grupo de pacientes selecionado
Pacientes assintomáticos não exigem tratamento.[99]Kaplan P, Baris H, De Meirleir L, et al. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr. 2013 Apr;172(4):447-58. http://www.ncbi.nlm.nih.gov/pubmed/22772880?tool=bestpractice.com No entanto, a terapia de reposição enzimática (TRE) deve ser considerada em todas as crianças sintomáticas, e em adultos com reduções significativas nas contagens sanguíneas (por exemplo, nível de hemoglobina <100 g/L [10 g/dL], plaquetas <100 x 10⁹/L), aumento significativo de órgãos (por exemplo, tamanho do baço >10x o normal), presença de doença esquelética demonstrada na ressonância nuclear magnética e/ou qualquer outro dano a órgãos (por exemplo, evidência de lesão do pulmão).[99]Kaplan P, Baris H, De Meirleir L, et al. Revised recommendations for the management of Gaucher disease in children. Eur J Pediatr. 2013 Apr;172(4):447-58. http://www.ncbi.nlm.nih.gov/pubmed/22772880?tool=bestpractice.com [100]Hughes D, Cappellini MD, Berger M, et al. Recommendations for the management of the haematological and onco-haematological aspects of Gaucher disease. Br J Haematol. 2007 Sep;138(6):676-86. https://onlinelibrary.wiley.com/doi/10.1111/j.1365-2141.2007.06701.x http://www.ncbi.nlm.nih.gov/pubmed/17655728?tool=bestpractice.com Os aspectos viscerais e esqueléticos da doença respondem bem à TRE, mas não há melhoras das manifestações neurológicas, pois a TRE não consegue atravessar a barreira hematoencefálica.[116]Vellodi A, Tylki-Szymanska A, Davies EH, et al. Management of neuronopathic Gaucher disease: revised recommendations. J Inherit Metab Dis. 2009 Oct;32(5):660-4. http://www.ncbi.nlm.nih.gov/pubmed/19655269?tool=bestpractice.com
A TRE foi associada a reações de hipersensibilidade graves, anafilaxia e reações relacionadas com à infusão. Considere a pré-medicação com um anti-histamínico, antipirético e/ou corticosteroide.
Opções primárias
imiglucerase: crianças e adultos: 60 unidades/kg por via intravenosa a cada 2 semanas inicialmente, ajustar de acordo com a resposta
ou
velaglucerase alfa: crianças com ≥4 anos de idade e adultos: 60 unidades/kg por via intravenosa a cada 2 semanas inicialmente, ajustar de acordo com a resposta
Doença de Fabry
terapia de suporte
Uma abordagem multidisciplinar é essencial, pois essas doenças são distúrbios multissistêmicos; muitos especialistas diferentes podem estar envolvidos no cuidado de pacientes individuais. É recomendável o envolvimento precoce de centros especializados; os cuidados devem ser coordenados pelo centro.
Há um grande número de aspectos gerais de suporte no manejo dessa doença multissistêmica.
terapia de reposição enzimática ou com chaperonas
Tratamento adicional recomendado para ALGUNS pacientes no grupo de pacientes selecionado
A produção de anticorpos é provocada pela TRE em homens, mas o impacto disso sobre a eficácia do tratamento é desconhecido. Mulheres não desenvolvem anticorpos, talvez porque sejam heterozigotas e tenham enzimas circulantes.
A TRE pode retardar a evolução da lesão orgânica nos rins e no coração, mas esses órgãos podem não retornar à função normal. Os homens devem ser tratados assim que forem diagnosticados; as mulheres devem ser tratadas se apresentarem sintomas de envolvimento de órgãos importantes.[67]Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. 2018 Apr;123(4):416-27. https://www.sciencedirect.com/science/article/pii/S1096719217307680 http://www.ncbi.nlm.nih.gov/pubmed/29530533?tool=bestpractice.com O início da TRE deve ser considerado em pacientes assintomáticas do sexo feminino com evidências laboratoriais, histológicas ou de imagem de envolvimento renal, cardíaco ou do sistema nervoso central.[67]Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. 2018 Apr;123(4):416-27. https://www.sciencedirect.com/science/article/pii/S1096719217307680 http://www.ncbi.nlm.nih.gov/pubmed/29530533?tool=bestpractice.com
A alfa-agalsidase e a beta-agalsidase são efetivas na prevenção de complicações renais e cardiovasculares, em comparação com a ausência de tratamento. A beta-agalsidase está associada a um menor risco de complicações cerebrovasculares, em comparação com a beta-agalsidase ou a ausência de tratamento.[134]El Dib R, Gomaa H, Ortiz A, et al. Enzyme replacement therapy for Anderson-Fabry disease: a complementary overview of a Cochrane publication through a linear regression and a pooled analysis of proportions from cohort studies. PLoS One. 2017 Mar 15;12(3):e0173358. https://journals.plos.org/plosone/article?id=10.1371/journal.pone.0173358 http://www.ncbi.nlm.nih.gov/pubmed/28296917?tool=bestpractice.com A TRE é eficaz na redução dos escores de dor e das concentrações de globotriaosilceramida no plasma, rim e coração.[122]El Dib R, Gomaa H, Carvalho RP, et al. Enzyme replacement therapy for Anderson-Fabry disease. Cochrane Database Syst Rev. 2016 Jul 25;(7):CD006663. http://onlinelibrary.wiley.com/doi/10.1002/14651858.CD006663.pub4/full http://www.ncbi.nlm.nih.gov/pubmed/27454104?tool=bestpractice.com
Recomendações sobre o uso de TRE se houver falta dessa terapia foram publicadas.[170]Linthorst GE, Germain DP, Hollak CE, et al; European Medicines Agency. Expert opinion on temporary treatment recommendations for Fabry disease during the shortage of enzyme replacement therapy (ERT). Mol Genet Metab. 2011 Jan;102(1):99-102. http://www.ncbi.nlm.nih.gov/pubmed/21123099?tool=bestpractice.com Essas recomendações foram publicadas em 2011, quando a beta-agalsidase (um dos dois produtos disponíveis para a doença de Fabry) estava passando por dificuldades de produção.
A pegunigalsidase alfa é outra opção para tratar a doença de Fabry em adultos, e tem a vantagem de uma meia-vida plasmática mais longa.[135]National Institute for Health and Care Excellence. Pegunigalsidase alfa for treating Fabry disease. Oct 2023 [internet publication]. https://www.nice.org.uk/guidance/ta915
A TRE foi associada a reações de hipersensibilidade graves, anafilaxia e reações relacionadas com à infusão. Considere a pré-medicação com um anti-histamínico, antipirético e/ou corticosteroide.
O migalastat é uma chaperona oral que aumenta a atividade da enzima endógena alfagalactosidase A em pacientes com uma mutação favorável. Ensaios demonstraram a segurança e a utilidade desta terapia em pacientes com mutações favoráveis, e o tratamento é atualmente licenciado e está disponível nos EUA, Europa, Canadá, Japão e vários outros países.[137]Germain DP, Hughes DA, Nicholls K, et al. Treatment of Fabry's disease with the pharmacologic chaperone migalastat. N Engl J Med. 2016 Aug 11;375(6):545-55. http://www.nejm.org/doi/full/10.1056/NEJMoa1510198 http://www.ncbi.nlm.nih.gov/pubmed/27509102?tool=bestpractice.com [138]National Institute for Health and Care Excellence. Migalastat for treating Fabry disease. Feb 2017 [internet publication]. https://www.nice.org.uk/guidance/hst4 [139]Hughes DA, Nicholls K, Shankar SP, et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J Med Genet. 2017 Apr;54(4):288-96. [Erratum in: J Med Genet. 2018.] https://jmg.bmj.com/content/54/4/288.long http://www.ncbi.nlm.nih.gov/pubmed/27834756?tool=bestpractice.com [140]Schiffmann R, Bichet DG, Jovanovic A, et al. Migalastat improves diarrhea in patients with Fabry disease: clinical-biomarker correlations from the phase 3 FACETS trial. Orphanet J Rare Dis. 2018 Apr 27;13(1):68. https://ojrd.biomedcentral.com/articles/10.1186/s13023-018-0813-7 http://www.ncbi.nlm.nih.gov/pubmed/29703262?tool=bestpractice.com Os médicos precisam confirmar que a mutação do paciente é favorável. O fabricante aconselha evitá-la se a taxa de filtração glomerular estimada (TFGe) for inferior a 30 mL/minute/1.73 m². O migalastate é adequado para pacientes sem experiência prévia e para pacientes que estão mudando de TRE.[141]Bichet DG, Hopkin RJ, Aguiar P, et al. Consensus recommendations for the treatment and management of patients with Fabry disease on migalastat: a modified Delphi study. Front Med (Lausanne). 2023 Sep 1:10:1220637. https://www.frontiersin.org/journals/medicine/articles/10.3389/fmed.2023.1220637/full http://www.ncbi.nlm.nih.gov/pubmed/37727761?tool=bestpractice.com
Opções primárias
alfa-agalsidase: crianças ≥7 anos de idade e adultos: 0.2 mg/kg por via intravenosa a cada 2 semanas
ou
beta-agalsidase: crianças ≥8 anos de idade e adultos: 1 mg/kg por via intravenosa a cada 2 semanas
ou
pegunigalsidase alfa: adultos: 1 mg/kg por via intravenosa a cada 2 semanas
ou
migalastat: adultos: 123 mg por via oral uma vez ao dia em dias alternados
profilaxia de ataque isquêmico transitório (AIT) e acidente vascular cerebral (AVC)
Tratamento recomendado para TODOS os pacientes no grupo de pacientes selecionado
O AVC e o ataque isquêmico transitório (AIT) requerem cuidadosas medidas preventivas primárias e secundárias.
alívio da dor
Tratamento recomendado para TODOS os pacientes no grupo de pacientes selecionado
O alívio da dor com gabapentina ou pregabalina é indicado para dor neuropática. Carbamazepina também é amplamente usada.[67]Ortiz A, Germain DP, Desnick RJ, et al. Fabry disease revisited: management and treatment recommendations for adult patients. Mol Genet Metab. 2018 Apr;123(4):416-27. https://www.sciencedirect.com/science/article/pii/S1096719217307680 http://www.ncbi.nlm.nih.gov/pubmed/29530533?tool=bestpractice.com [117]Wanner C, Arad M, Baron R, et al. European expert consensus statement on therapeutic goals in Fabry disease. Mol Genet Metab. 2018 Jul;124(3):189-203. http://www.ncbi.nlm.nih.gov/pubmed/30017653?tool=bestpractice.com
Anti-inflamatórios não esteroidais devem ser usados com moderação, pois esses pacientes muitas vezes apresentam nefropatia.
Opções primárias
gabapentina: crianças: consulte um especialista para obter orientação quanto à dose; adultos: 1800-3600 mg/dia por via oral administrados em 3-4 doses fracionadas
ou
pregabalina: crianças: consulte um especialista para obter orientação quanto à dose; adultos: 150-300 mg/dia por via oral administrados em 3 doses fracionadas
ou
carbamazepina: crianças: consulte um especialista para obter orientação quanto à dose; adultos: 200-1600 mg/dia por via oral administrados em 3-4 doses fracionadas
cirurgia cosmética ou tratamento a laser
Tratamento recomendado para TODOS os pacientes no grupo de pacientes selecionado
As lesões cutâneas podem necessitar de cirurgia estética ou laserterapia. [Figure caption and citation for the preceding image starts]: Lesões cutâneas na doença de Fabry: (A) palmas das mãos, (B) lábios, (C) mucosa labialOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; usado com permissão [Citation ends].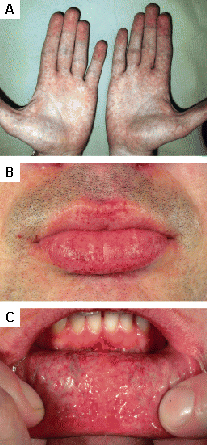 [Figure caption and citation for the preceding image starts]: Lesões cutâneas na doença de Fabry: (A) flanco, (B) genitais, (C) umbigo, (D) coluna lombar, (E) pododáctilosOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; usado com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Lesões cutâneas na doença de Fabry: (A) flanco, (B) genitais, (C) umbigo, (D) coluna lombar, (E) pododáctilosOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; usado com permissão [Citation ends].
inibidor da enzima conversora da angiotensina (IECA) ou antagonista do receptor de angiotensina II
Tratamento recomendado para TODOS os pacientes no grupo de pacientes selecionado
Pressão arterial e proteinúria devem ser monitoradas regularmente com intervenção precoce.
Opções primárias
enalapril: crianças: consulte um especialista para obter orientação quanto à dose; adultos: 2.5 a 40 mg por via oral uma vez ao dia
ou
irbesartana: crianças: consulte um especialista para obter orientação quanto à dose; adultos: 150-300 mg por via oral uma vez ao dia
marca-passo + avaliação para cirurgia cardíaca
Tratamento recomendado para TODOS os pacientes no grupo de pacientes selecionado
A avaliação cardíaca é essencial nos pacientes com doença de Fabry. O reconhecimento precoce da arritmia é importante, e ela deve ser tratada com um marca-passo. Além disso, cirurgias, incluindo inserção de marca-passos, ressecção do septo, substituição de valva, e até mesmo o transplante cardíaco, devem ser consideradas.[119]Linhart A, Elliott PM. The heart in Anderson-Fabry disease and other lysosomal storage diseases. Heart. 2007 Apr;93(4):528-35. http://www.ncbi.nlm.nih.gov/pubmed/17401074?tool=bestpractice.com
mucopolissacaridose (MPS)
terapia de suporte
Uma abordagem multidisciplinar é essencial, pois essas doenças são distúrbios multissistêmicos; muitos especialistas diferentes podem estar envolvidos no cuidado de pacientes individuais. É recomendável o envolvimento precoce de centros especializados; os cuidados devem ser coordenados pelo centro.
Problemas musculoesqueléticos incluem deformidade espinhal, síndrome do túnel do carpo e neuropatia compressiva. Pode haver compressão da medula espinhal decorrente de estenose na região craniocervical.[145]Solanki GA, Alden TD, Burton BK, et al. A multinational, multidisciplinary consensus for the diagnosis and management of spinal cord compression among patients with mucopolysaccharidosis VI. Mol Genet Metab. 2012 Sep;107(1-2):15-24. http://www.sciencedirect.com/science/article/pii/S1096719212002740 http://www.ncbi.nlm.nih.gov/pubmed/22938833?tool=bestpractice.com Hidrocefalia comunicante e convulsões podem ocorrer. Os pacientes também podem apresentar valvopatia cardíaca que requer avaliação cirúrgica.
Todos esses distúrbios necessitam de avaliação detalhada. Fisioterapia, suportes, injeções locais, tudo é possível; terapia cirúrgica pode ser necessária.[144]van der Linden MH, Kruyt MC, Sakkers RJ, et al. Orthopaedic management of Hurler's disease after hematopoietic stem cell transplantation: a systematic review. J Inherit Metab Dis. 2011 Jun;34(3):657-69. http://link.springer.com/article/10.1007/s10545-011-9304-x/fulltext.html http://www.ncbi.nlm.nih.gov/pubmed/21416194?tool=bestpractice.com [145]Solanki GA, Alden TD, Burton BK, et al. A multinational, multidisciplinary consensus for the diagnosis and management of spinal cord compression among patients with mucopolysaccharidosis VI. Mol Genet Metab. 2012 Sep;107(1-2):15-24. http://www.sciencedirect.com/science/article/pii/S1096719212002740 http://www.ncbi.nlm.nih.gov/pubmed/22938833?tool=bestpractice.com
terapia de reposição enzimática
Tratamento recomendado para TODOS os pacientes no grupo de pacientes selecionado
A terapia de reposição enzimática (TRE) tem benefício estabelecido na MPS I, II, IVA (síndrome de Morquio tipo A), VI e tipo VII (síndrome de Sly).[148]Hendriksz CJ, Burton B, Fleming TR, et al.; STRIVE Investigators. Efficacy and safety of enzyme replacement therapy with BMN 110 (elosulfase alfa) for Morquio A syndrome (mucopolysaccharidosis IVA): a phase 3 randomised placebo-controlled study. J Inherit Metab Dis. 2014 Nov;37(6):979-90. http://link.springer.com/article/10.1007/s10545-014-9715-6/fulltext.html http://www.ncbi.nlm.nih.gov/pubmed/24810369?tool=bestpractice.com [149]Brunelli MJ, Atallah ÁN, da Silva EM. Enzyme replacement therapy with galsulfase for mucopolysaccharidosis type VI. Cochrane Database Syst Rev. 2021 Sep 17;(9):CD009806. https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD009806.pub3/full http://www.ncbi.nlm.nih.gov/pubmed/34533215?tool=bestpractice.com [150]Jameson E, Jones S, Remmington T. Enzyme replacement therapy with laronidase (Aldurazyme®) for treating mucopolysaccharidosis type I. Cochrane Database Syst Rev. 2019 Jun 18;6(6):CD009354. https://www.cochranelibrary.com/cdsr/doi/10.1002/14651858.CD009354.pub5/full http://www.ncbi.nlm.nih.gov/pubmed/31211405?tool=bestpractice.com [151]Wikman-Jorgensen PE, López Amorós A, Peris García J, et al. Enzyme replacement therapy for the treatment of Hunter disease: a systematic review with narrative synthesis and meta-analysis. Mol Genet Metab. 2020 Sep - Oct;131(1-2):206-10. http://www.ncbi.nlm.nih.gov/pubmed/32773276?tool=bestpractice.com [152]Gomes DF, Gallo LG, Leite BF, et al. Clinical effectiveness of enzyme replacement therapy with galsulfase in mucopolysaccharidosis type VI treatment: systematic review. J Inherit Metab Dis. 2019 Jan;42(1):66-76. http://www.ncbi.nlm.nih.gov/pubmed/30740728?tool=bestpractice.com Várias enzimas estão aprovadas para essas indicações. No Reino Unido, o National Institute for Health and Care Excellence recomenda a elosulfase alfa como uma opção para o tratamento da MPS IVA para pessoas de todas as idades, e está disponível apenas sob um acordo comercial.[153]National Institute for Health and Care Excellence. Elosulfase alfa for treating mucopolysaccharidosis type 4A: highly specialised technologies guidance. Apr 2022 [internet publication]. https://www.nice.org.uk/guidance/hst19 Estão em andamento ensaios clínicos de Fase 3 com a vestronidase alfa.[154]NHS; National Institute for Health Research (NIHR). NIHR Innovation Observatory evidence briefing. Vestronidase alfa (UX-003) for mucopolysaccharidosis type VII (MPS 7; Sly syndrome) NIHRIO (HSRIC) ID: 11463. Apr 2017 [internet publication]. http://www.io.nihr.ac.uk/wp-content/uploads/migrated_new/11463-Vestronidase-alfa-UX-003.pdf [155]ClinicalTrials.gov. A study of UX003 recombinant human beta-glucuronidase (rhGUS) enzyme replacement therapy in subjects with mucopolysaccharidosis type 7, Sly syndrome (MPS 7). Jul 2020 [internet publication]. https://clinicaltrials.gov/ct2/show/NCT02432144
A TRE foi associada a reações de hipersensibilidade graves, anafilaxia e reações relacionadas com à infusão. Considere a pré-medicação com um anti-histamínico, antipirético e/ou corticosteroide.
Opções primárias
MPS I
laronidase: crianças ≥5 anos de idade e adultos: 0.58 mg/kg por via intravenosa uma vez por semana
ou
MPS II
idursulfase: crianças ≥5 anos de idade e adultos: 0.5 mg/kg por via intravenosa uma vez por semana
ou
síndrome de Morquio tipo A (IVA) na MPS
elosulfase alfa: crianças ≥5 anos de idade e adultos: 2 mg/kg por via intravenosa uma vez por semana
ou
MPS VI
galsulfase: crianças ≥5 anos de idade e adultos: 1 mg/kg por via intravenosa uma vez por semana
ou
MPS VII
vestronidase alfa: crianças e adultos: 4 mg/kg por via intravenosa a cada 2 semanas
transplante de células-tronco
Tratamento adicional recomendado para ALGUNS pacientes no grupo de pacientes selecionado
O transplante de células-tronco deve ser considerado para os pacientes gravemente afetados e tem valor estabelecido, por exemplo, na MPS I e na MPS VI graves.[147]Boelens JJ. Trends in hematopoietic stem cell transplantation for inborn errors of metabolism. J Inherit Metab Dis. 2006 Apr-Jun;29(2-3):413-20. http://www.ncbi.nlm.nih.gov/pubmed/16763911?tool=bestpractice.com
Doença de Pompe
terapia de suporte
Uma abordagem multidisciplinar é essencial, pois essas doenças são distúrbios multissistêmicos; muitos especialistas diferentes podem estar envolvidos no cuidado de pacientes individuais. É recomendável o envolvimento precoce de centros especializados; os cuidados devem ser coordenados pelo centro.
Os cuidados gerais e de suporte para neonatos são multidisciplinares, envolvendo neurologistas, anestesistas e cardiologistas.
Insuficiência respiratória e apneia obstrutiva do sono podem necessitar de diferentes graus de suporte ventilatório.
terapia de reposição enzimática
Tratamento adicional recomendado para ALGUNS pacientes no grupo de pacientes selecionado
A terapia de reposição enzimática (TRE) é licenciada em crianças e adultos. A avalglucosidase é uma opção para a doença de Pompe de início tardio em crianças com 1 ano de idade ou mais.
A cipaglucosidase alfa é outra opção aprovada para o tratamento da doença de Pompe de início tardio em adultos que não estiverem melhorando com a TRE atual. A cipaglucosidase alfa só está aprovada para uso em combinação com o miglustate (um estabilizador enzimático).[165]Blair HA. Cipaglucosidase alfa: first approval. Drugs. 2023 Jun;83(8):739-45. https://link.springer.com/article/10.1007/s40265-023-01886-5 http://www.ncbi.nlm.nih.gov/pubmed/37184753?tool=bestpractice.com
As diretrizes do consenso europeu recomendam um período inicial de 2 anos de TRE para adultos sintomáticos. A TRE pode ser mantida durante a gravidez e lactação. O músculo esquelético e a função respiratória devem ser avaliados durante o tratamento.[166]van der Ploeg AT, Kruijshaar ME, Toscano A, et al. European consensus for starting and stopping enzyme replacement therapy in adult patients with Pompe disease: a 10-year experience. Eur J Neurol. 2017 Jun;24(6):768-e31. http://www.ncbi.nlm.nih.gov/pubmed/28477382?tool=bestpractice.com
A TRE foi associada a reações de hipersensibilidade graves, anafilaxia e reações relacionadas com à infusão. Considere a pré-medicação com um anti-histamínico, antipirético e/ou corticosteroide.
Opções primárias
alglucosidase alfa: crianças e adultos: 20 mg/kg por via intravenosa a cada 2 semanas
ou
avalglucosidase alfa: crianças ≥1 ano de idade e <30 kg: 40 mg/kg por via intravenosa a cada 2 semanas; crianças ≥1 ano de idade e ≥30 kg e adultos: 20 mg/kg por via intravenosa a cada 2 semanas
ou
cipaglucosidase alfa: adultos ≥40 kg de peso corporal: 20 mg/kg por via intravenosa a cada 2 semanas
Mais cipaglucosidase alfaComeçar 2 semanas após a última dose da terapia de reposição enzimática. Administrar 1 hora após a dose de miglustate (em no máximo 3 horas após a dose do miglustate).
e
miglustate: adultos ≥40 kg a <50 kg de peso corporal: 195 mg por via oral a cada 2 semanas; adultos ≥50 kg de peso corporal: 260 mg por via oral a cada 2 semanas
Mais miglustateComeçar 2 semanas após a última dose da terapia de reposição enzimática. Administrar 1 hora antes da dose de cipaglucosidase alfa.
Existem várias marcas de miglustate disponíveis. Opfolda® é a marca aprovada para uso na doença de Pompe, e deve ser usada para esta indicação.
doença de Tay-Sachs
terapia de suporte
Somente cuidados paliativos são necessários para a forma infantil.
As formas de início na fase juvenil ou adulta requerem cuidados de suporte, educação quanto a necessidades especiais e avaliação neurológica. Demência e ataxia são complicações em longo prazo que precisam ser consideradas no manejo.
A terapia de reposição enzimática não está disponível para a doença de Tay-Sachs.
Doença de Niemann-Pick
terapia de suporte
Cuidados paliativos são necessários apenas para a forma infantil grave de Niemann-Pick do tipo A. Os cuidados de suporte são indicados para as formas menos graves.
Niemann-Pick do tipo B se apresenta tipicamente em adultos com doença pulmonar e/ou hepatoesplenomegalia.
Niemann-Pick do tipo C é extremamente variável, mas a avaliação neurológica geralmente é importante. A terapia de reposição enzimática está disponível para o tipo C.
terapia de redução de substrato
Tratamento adicional recomendado para ALGUNS pacientes no grupo de pacientes selecionado
A terapia de redução de substrato (TRS) está apenas disponível para Niemann-Pick do tipo C.[54]Patterson MC, Clayton P, Gissen P, et al. Recommendations for the detection and diagnosis of Niemann-Pick disease type C: an update. Neurol Clin Pract. 2017 Dec;7(6):499-511. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5800709 http://www.ncbi.nlm.nih.gov/pubmed/29431164?tool=bestpractice.com A TRS com miglustate demonstrou estabilizar os sintomas neurodegenerativos, incluindo disfagia.[168]Pineda M, Walterfang M, Patterson MC. Miglustat in Niemann-Pick disease type C patients: a review. Orphanet J Rare Dis. 2018 Aug 15;13(1):140. https://ojrd.biomedcentral.com/articles/10.1186/s13023-018-0844-0 http://www.ncbi.nlm.nih.gov/pubmed/30111334?tool=bestpractice.com
Opções primárias
miglustate: adultos: 100 mg por via oral três vezes ao dia, ajustar de acordo com a resposta
Mais miglustateExistem várias marcas de miglustate disponíveis; certifique-se de usar a marca correta para essa indicação.
Escolha um grupo de pacientes para ver nossas recomendações
Observe que as formulações/vias e doses podem diferir entre nomes e marcas de medicamentos, formulários de medicamentos ou localidades. As recomendações de tratamento são específicas para os grupos de pacientes. Ver aviso legal
O uso deste conteúdo está sujeito ao nosso aviso legal