Etiologia
Doenças do armazenamento lisossomal (DALs) são distúrbios em um único gene, geralmente decorrentes de defeitos hereditários na síntese (raramente na ativação defeituosa, no tráfego intracelular ou no direcionamento) de enzimas lisossomais.[2]
Essas enzimas são essenciais para a hidrólise ácida de macromoléculas extracelulares (por exemplo, glicofingolipídeos, derivados da decomposição de membranas de células sanguíneas, gangliosídeos derivados de neurônios e glicosaminoglicanos derivados de tecido conjuntivo e matriz extracelular). Substrato substancial também deriva da renovação intracelular de macromoléculas.
A maioria das DALs é autossômica recessiva; no entanto, 3 das doenças (Fabry, mucopolissacaridose [MPS] II e Danon) são ligadas ao sexo. Há uma considerável heterogeneidade fenotípica entre todas as DALs, e as correlações genótipo-fenótipo geralmente são insatisfatórias. Contudo, em geral, quanto menor a atividade enzimática residual, maior o acúmulo de substrato, menor a idade de início e mais grave a doença.[1][21]
As deleções genes, mutações sem sentido e mutações que interferem intensamente na estrutura terciária da enzima geralmente são incompatíveis com a vida (por exemplo, como observado na doença de Gaucher do tipo 2) ou associadas a manifestações graves. As mutações associadas a atividade enzimática residual têm um nível elevado de heterogeneidade fenotípica, e até irmãos com a mesma mutação podem ter manifestações clínicas amplamente divergentes.
O envolvimento neurológico na doença de Gaucher não é observado nos pacientes que têm pelo menos um alelo com a mutação N370S (comum em judeus asquenazes), pois isso é suficiente para uma atividade enzimática residual que "protege" o sistema nervoso central.[22][23]
Esfingolipidoses
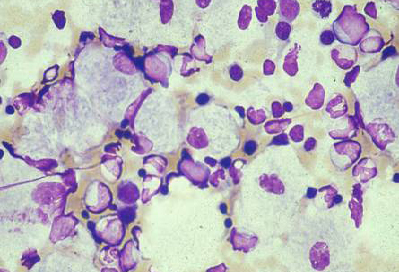
Doença de Gaucher (deficiência de beta-glucocerebrosidase) do tipo 1 (forma mais comum), dos tipos 2 e 3 (com envolvimento do SNC)[Figure caption and citation for the preceding image starts]: Aspirado da medula óssea mostrando célula de Gaucher típica. Este é um macrófago que ingeriu material celular; o substrato não degradado (glicosilceramida) se acumula nos lisossomosDo acervo pessoal do professor Atul B. Mehta [Citation ends].
 [Figure caption and citation for the preceding image starts]: Ressonância nuclear magnética do esqueleto na doença de Gaucher do tipo 1 mostrando deposição esquelética disseminada de substrato com necrose associada e infarto ósseo. Há necrose avascular da cabeça femoral (seta)Do acervo pessoal do professor Atul B. Mehta [Citation ends].
[Figure caption and citation for the preceding image starts]: Ressonância nuclear magnética do esqueleto na doença de Gaucher do tipo 1 mostrando deposição esquelética disseminada de substrato com necrose associada e infarto ósseo. Há necrose avascular da cabeça femoral (seta)Do acervo pessoal do professor Atul B. Mehta [Citation ends].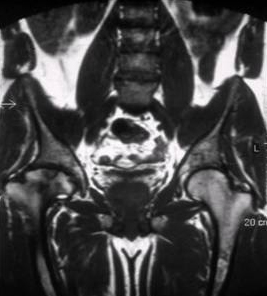
Doença de Fabry (deficiência de alfagalactosidase)[Figure caption and citation for the preceding image starts]: Imagem de microscópio eletrônico de biópsia de células epiteliais pulmonares na doença de Fabry mostrando depósitos característicos de substrato em lisossomos, formando "corpos em zebra": (A) aumento de 8000x, (B) aumento de 62,500xKelly MM, Leigh R, McKenzie R, et al. Induced sputum examination: diagnosis of pulmonary involvement in Fabry's disease. Thorax. 2000 Aug;55(8):720-1; usado com permissão [Citation ends].
 [Figure caption and citation for the preceding image starts]: Lesões cutâneas na doença de Fabry: (A) palmas das mãos, (B) lábios, (C) mucosa labialOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; usado com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Lesões cutâneas na doença de Fabry: (A) palmas das mãos, (B) lábios, (C) mucosa labialOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; usado com permissão [Citation ends].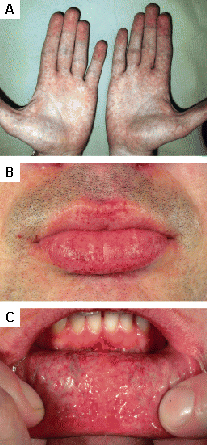 [Figure caption and citation for the preceding image starts]: Lesões cutâneas na doença de Fabry: (A) flanco, (B) genitais, (C) umbigo, (D) coluna lombar, (E) pododáctilosOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; usado com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Lesões cutâneas na doença de Fabry: (A) flanco, (B) genitais, (C) umbigo, (D) coluna lombar, (E) pododáctilosOrteu CH, Jansen T, Lidove O, et al. Fabry disease and the skin: data from FOS, the Fabry Outcome Survey. Br J Dermatol. 2007 Aug;157(2):331-7; usado com permissão [Citation ends].
Tay-Sachs (deficiência de beta hexosaminidase A)
Sandhoff (deficiência de beta hexosaminidase B)
Krabbe (deficiência de galactoceramida betagalactosidase)
Niemann-Pick tipos A, B (deficiência de esfingomielinase ácida)
Niemann-Pick tipo C (defeito complexo no transporte)
Leucodistrofia metacromática (deficiência arilsulfatase A)
Gangliosidose GM1 (deficiência de beta-galactosidase)
Mucopolissacaridose (MPS)
Forma grave de MPS I (deficiência de alfa-L-iduronidase) (Hurler), formas mais leves (Hurler-Scheie, Scheie)
MPS II/síndrome de Hunter (deficiência de iduronato-2-sulfatase).
MPS III/síndrome de Sanfilippo (4 subtipos: A, B, C, D) (deficiência de heparina-N-sulfatase e outras enzimas)
MPS IV/síndrome de Morquio (2 subtipos: A, B) (deficiência de N-acetilgalactosamina 6-sulfatase e betagalactosidase)
MPS VI/síndrome Maroteaux-Lamy (deficiência de N-acetilgalactosamina 4-sulfatase)
MPS VII/síndrome de Sly (deficiência de beta-glucuronidase)
MPS IX/síndrome de Natowicz (deficiência de hialuronidase)
MPS X/Deficiência de arilsulfatase K
Outra
Doença de Pompe/doença de depósito de glicogênio do tipo 2 (deficiência de maltase ácida).
Glicoproteinoses (por exemplo, aspartilglicosaminúria, fucosidose, alfa e beta-manosidose, doença de Schindler)
Deficiência de sulfatase múltipla, mucolipidose II, III, IV, doença de Danon, doença de Wolman, defeitos de transporte lisossomal (por exemplo, cistinose, doença do armazenamento de ácido siálico)
Fisiopatologia
A deficiência de enzima perturba o metabolismo intermediário com acúmulo de substrato não degradado.[1][2] Isso causa não apenas o aumento de células e órgãos individuais, mas também causa anormalidades estruturais e bioquímicas secundárias. Ocorre hipertrofia lisossomal, que causa grande perturbação na função lisossomal.
As alterações neuronais são parcialmente estruturais e decorrentes do aumento do armazenamento; porém, foi demonstrada apoptose acelerada de neurônios nos distúrbios de mucopolissacaridose; além disso, foi demonstrada uma perturbação do metabolismo de cálcio, resultando em morte celular neuronal excessiva na doença de Gaucher neuronopática.[24][25]
O acúmulo extralisossomal de substrato pode prejudicar a função da membrana, o metabolismo mitocondrial, os canais iônicos de membranas e transportadores, e a transdução de sinal através de microdomínios (lipid rafts) da membrana.[1][26]
Demonstrou-se ativação macrofágica, possivelmente mediada pela liberação de citocinas, na doença de Gaucher e em outras doenças do armazenamento lisossomal (DALs).[27][28] O acúmulo de lisosfingolipídeos (isto é, glicoesfingolipídeos que não possuem ácidos graxos N-acilados) pode ter significância patogênica nas doenças de Krabbe, Gaucher e Fabry.[29]
Fisiopatologia específica da doença
Doença de Gaucher: devida à deficiência de beta-glucocerebrosidase. Isso torna o lisossoma incapaz de quebrar os glicofingolipídeos, levando a um acúmulo de glicosilceramidas.[30]
Doença de Fabry: causada pela deficiência de alfagalactosidase, que resulta no acúmulo de globotriaosilceramida nos lisossomas.[31]
Doença de Tay-Sachs: resulta da deficiência de beta-hexosaminidase A. Os gangliosídeos se acumulam nos lisossomos, principalmente nos neurônios.[32]
A doença de Niemann-Pick (DNP): tipos A e B é decorrente da deficiência de esfingomielinase ácida. A DNP do tipo A é um distúrbio neurodegenerativo grave que tipicamente se apresenta na primeira infância, enquanto a DNP do tipo B, que geralmente preserva o sistema nervoso, é caracterizada por acúmulo de substrato no sistema reticuloendotelial (causando hepatoesplenomegalia, doença pulmonar) e se apresenta mais tarde. A DNP do tipo C é um distúrbio complexo de transporte intracelular e processamento de colesterol, que geralmente se apresenta na infância com ataxia, movimentos oculares desordenados e dificuldades de aprendizagem, mas pode se manifestar em adultos como uma doença neurológica ou com hepatoesplenomegalia.[33]
Mucopolissacaridoses: caracterizadas por degradação incompleta e acúmulo progressivo de glicosaminoglicanos.[34]
Doença de Pompe: causada pela deficiência de maltase ácida, levando ao acúmulo de glicogênio nos lisossomas.[35]
Classificação
Esfingolipidoses
Incidência: >1/50,000 pessoas[5]
Doença de Gaucher (deficiência de beta-glucocerebrosidase) do tipo 1 (forma mais comum), dos tipos 2 e 3 (com envolvimento do sistema nervoso central)
Doença de Fabry (deficiência de alfagalactosidase)
Incidência: <1/100,000 pessoas[5]
Tay-Sachs (deficiência de beta hexosaminidase A)
Sandhoff (deficiência de beta hexosaminidase B)
Krabbe (deficiência de galactoceramida betagalactosidase)
Niemann-Pick tipos A, B (deficiência de esfingomielinase ácida)
Niemann-Pick tipo C (defeito complexo no transporte)
Leucodistrofia metacromática (deficiência arilsulfatase A)
Gangliosidose GM1 (deficiência de beta-galactosidase)
Mucopolissacaridose (MPS)
Incidência: >1/100,000 pessoas[5]
Forma grave de MPS I (deficiência de alfa-L-iduronidase) (Hurler), formas mais leves (Hurler-Scheie, Scheie)
MPS II/síndrome de Hunter (deficiência de iduronato-2-sulfatase)
Incidência: <1/100,000 pessoas[5]
MPS III/síndrome de Sanfilippo (4 subtipos: A, B, C, D) (deficiência de heparina-N-sulfatase e outras enzimas)
MPS IV/síndrome de Morquio (2 subtipos: A, B) (deficiência de N-acetilgalactosamina 6-sulfatase e betagalactosidase)
MPS VI/síndrome Maroteaux-Lamy (deficiência de N-acetilgalactosamina 4-sulfatase)
MPS VII/síndrome de Sly (deficiência de beta-glucuronidase)
MPS IX/síndrome de Natowicz (deficiência de hialuronidase)
MPS X/Deficiência de arilsulfatase K
Outra
Incidência: >1/50,000 pessoas[5]
Doença de Pompe/doença de depósito de glicogênio do tipo 2 (deficiência de maltase ácida)
Incidência: >1/100,000 pessoas[5]
Glicoproteinoses (por exemplo, aspartilglicosaminúria, fucosidose, alfa e beta-manosidose, doença de Schindler)
Deficiência de sulfatase múltipla, mucolipidose II, III, IV, doença de Danon, doença de Wolman, defeitos de transporte lisossomal (por exemplo, cistinose, doença do armazenamento de ácido siálico)
O uso deste conteúdo está sujeito ao nosso aviso legal