Approach
A full history and clinical examination are the first steps towards diagnosis. Carer reports of changes in behavioural and psychological symptoms are the main source of information for clinicians, and should be assessed through carer interviews.[72] When a generalist suspects frontotemporal dementia (FTD) in an adult aged <70 years, or whenever there is a family history of dementia, referral should be made to a memory assessment specialist or service for a comprehensive assessment.[73]
Diagnosing FTD remains a challenge, and the condition is underdiagnosed. This is in part because of symptom overlap between behavioural variant FTD (bvFTD) and a number of primary psychiatric disorders (PPDs).[28][74] Despite the overlapping features, there are clinical elements that can differentiate between bvFTD and PPDs.[75]
Using an algorithmic approach may aid the process of systematically ruling in or out primary psychiatric conditions.[76]
The Neuropsychiatric International Consortium for Frontotemporal Dementia has proposed a list of recommendations for the assessment of bvFTD to enhance its differentiation from PPDs.[28][77]
[Figure caption and citation for the preceding image starts]: Clinical features aiding the differential diagnosis between behavioural variant FTD (bvFTD) and primary psychiatric illnessAntonioni A et al. Ex Rev of Neuro 2025; 25(3): 323-57; used with permission of Informa UK Limited, trading as Taylor & Francis Group, www.tandfonline.com [Citation ends].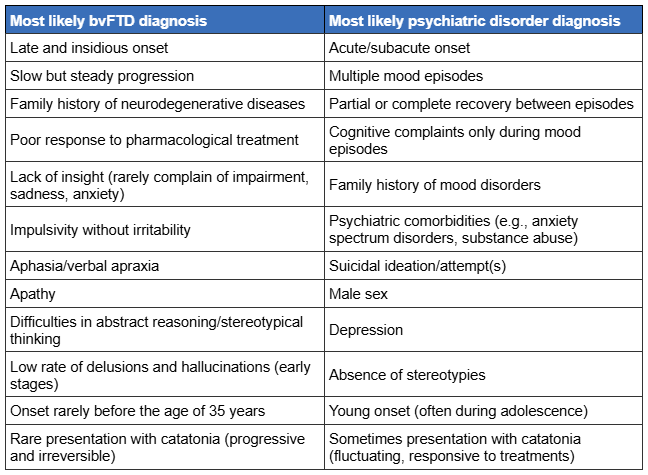
History
A full history should be obtained independently from a person who was familiar with the patient before disease onset. A thorough account is required of any changes in the patient's social interpersonal conduct, regulation of personal conduct (e.g., disinhibition), emotions (e.g., apathy, empathy), stereotypical behaviours, eating habits, cognitive abilities, and awareness about their illness, as well as enduring aspects of temperament, character, attitudes, and habits.[72]
Common features at onset are:
Insidious onset of coarsened personality, and changes in social behaviour and habits
Gradually progressive loss of language fluency or comprehension
Progressive self-neglect and abandonment of work, activities, and social contacts.
It is essential to establish a chronology of symptoms. In FTD, changes in personality, language, habits, and activity generally precede the development of memory impairment, disorientation, or apraxias. Most patients with FTD will exhibit one or more behavioural and psychological symptoms of dementia, such as apathy, disinhibition, appetite and eating abnormalities, irritability, agitation and aggression, aberrant motor behaviour, sleep issues, and wandering, during the course of the disease.[78][79] Disinhibition may present as impulsivity, loss of etiquette, excessive or perseverative actions, sexual inappropriateness and hypersexuality, and other transgressive behaviours such as shoplifting.[80][81] Psychosis and/or depression may be present but less frequently than in other types of dementia, and symptoms of depression may precede the onset of FTD.[78][79][82]
FTD may be associated with falls and parkinsonian symptoms. Such features are suggestive of FTD with parkinsonism, corticobasal syndrome, and progressive supranuclear palsy. FTD can also be associated with muscle wasting and weakness, which is suggestive of amyotrophic lateral sclerosis (ALS). Additionally, enquiring about motor symptoms such as weakness, clumsiness, mobility difficulties, tremors, spasms, poor balance, difficulty navigating the stairs, predisposition to falls, and swallowing issues may hint at parkinsonism (related to corticobasal syndrome [CBS] or progressive supranuclear palsy [PSP]) or ALS.
Since primary progressive aphasia (PPA) constitutes the other major group in the FTD spectrum, history taking should include any subjective or observed changes in language proficiency or communication.
A full family history is required if FTD is suspected. The Goldman score is a measure of strength of family history that takes account of degree of relativity within families.[83] A positive family history should extend beyond FTD and young-onset dementia to include Parkinson's disease and related disorders, ALS, and unexplained late-onset psychiatric disorders.[28]
The clinician should ask about previous episodes with symptoms of mental disturbance (whether treated or untreated), such as recurring mania or depression, which may point to a primary psychiatric disorder rather than FTD. Distressing compulsions are also unlikely to be due to FTD.
The possibility of conditions such as hyperthyroidism should be explored (by enquiring about, for example, heat intolerance, weight loss despite excessive eating, tremulousness, and palpitations) because features of hyperthyroidism, such as irritability, restlessness, increased eating (with decreased satiation), and distractibility, may result in a presentation that mimics FTD.
The careful documentation of disability forms the basis for treatment planning. Disabilities include disorientation, impaired communication, failures of self-care and socialisation, loss of bladder and bowel control, and inability to dress.
Examination
Patients with FTD may have no neurological abnormality or cognitive impairment at disease onset. Some patients present with subtle motor signs and/or have executive cognitive deficits at disease onset. Subtle signs suggestive of frontal lobe dysfunction arise as the disease progresses, including utilisation behaviour and one or more of glabellar, snout, sucking, rooting, or grasp reflexes. Many patients manifest parkinsonism, with some combination of tremor at rest, hypophonia, slowness of movement, narrowed range of facial expression, stooped posture, and unsteady gait. The presence of other motor signs such as dystonia, clonus, and alien limb should be ascertained. Similarly, oculomotor signs such as delayed vertical saccades and vertical nystagmus should be evaluated. ALS, when present, is characterised by progressive asymmetrical weakness of spinal or bulbar muscles, which may be accompanied by pharyngeal weakness. Various impairments in language, such as those related to articulation, fluency, grammar, comprehension, naming, repeating, semantics, reading, writing, word retrieval, and circumlocutions should be noted. Although not commonly a prominent feature of early stages of FTD, memory and other cognitive deficits such as agnosia and apraxia should also be observed.
Cognitive testing
Cognitive test performance should be recorded at initial assessment and then every 6 months. It is not unusual for cognitive test scores to be in the non-impaired range for up to 1 year after presentation and to decline steadily for about 3-4 years before the patient becomes untestable. These are given by specialists and neuropsychologists, and assist in the diagnosis by documenting a predominance of deficits in the executive and/or language domains of cognition, with relative sparing of memory, orientation, and praxis.
The mini-mental state examination (MMSE) remains the most widely used (and best understood) simple cognitive test. However, patients with FTD can score above the cut-off score of 24/30 even if they have significant cognitive impairment, so it is of limited usefulness for detecting FTD. Therefore, other cognitive tests are usually favoured in practice for FTD:
The Montreal Cognitive Assessment (MoCA) can be used to assess for decline in cognition over time; some subsets may give information about poor frontal lobe functioning and track progression.[84][85]
The Addenbrooke’s Cognitive Examination III (ACE III) is a multi-domain test of attention, memory, language, fluency, and visuospatial functions. Maximum total score is 100 with sub-scores for various domains also calculated. It has been validated as a screening tool for cognitive deficits in FTD and Alzheimer’s disease.[86]
The Frontal Assessment Battery (FAB) is a bedside test that can be administered in 10 minutes, and is helpful for screening and identifying frontal lobe dysfunction.[87]
The Executive Interview (EXIT) is a useful bedside screening tool for detecting possible frontal lobe dysfunction. It comprises a short screen of 25 items, and takes approximately 15 minutes.[88] The Quick EXIT is an abridged, 14-item version of the original EXIT that was developed by omitting 11 items that were assessed to fit the scale less well.[89]
The Frontal Behavioural Inventory (FBI) is a 24-item, quantifiable questionnaire completed by interviewing an informant.[90] It is aimed at identifying early behavioural and personality changes in bvFTD.[91]
The Cambridge Behavioural Inventory Revised (CBI-R) is an informant-based questionnaire used to evaluate behavioural symptoms in neurodegenerative diseases, including FTD.[92]
The Sydney Language Battery (SYDBAT) is a test to characterise language deficits in primary progressive aphasia (PPA) variants.[93][94] Four language subtests (naming, word comprehension, repetition, and semantic association) are assessed. A speech language therapist may be involved for a comprehensive assessment of language and devising interventions and compensatory strategies in patients with PPA.[95]
Neurobehavioural scales may also be of use in differentiating from other forms of dementia.[96]
The course of FTD differs from that of most other forms of dementia largely because memory problems are not severe in early stages. Most dementia rating scales are biased towards detection of worsening in Alzheimer's disease. The frontotemporal rating scale (FRS) was developed specifically for FTD, and can detect functional deterioration over 12 months.[97] The degree of dementia severity is more accurately estimated by the FRS than by conventional dementia rating scales.
The Ekman 60 faces test is a test used for checking facial emotion recognition.[98] Poor emotional processing is detectable in patients with FTD and is an important clinical feature in bvFTD and semantic dementia.[99] Emotion recognition can be significantly impaired in bvFTD in comparison to Alzheimer’s disease across all emotions other than happiness.[100] Additionally, recognition of anger is found to be more impaired in FTD than in Alzheimer's disease, while deficit in recognition of fear is more characteristic of Alzheimer's disease.[101]
More comprehensive neuropsychological testing of multiple domains is helpful, beyond the initial cognitive tests described earlier in this section, particularly in the early stages when the manifestations of FTD are subtle. Examples of formal test batteries that are appropriate in this setting include the Delis-Kaplan Executive Function System (D-KEFS), Executive and Social Cognition Battery (ESCB), and selected items from the Wechsler Adult Intelligence Scale (WAIS).[102]
Laboratory evaluation
Routine screening tests that should be done at presentation to exclude other diseases include the following:
FBC
CRP
Thyroid-stimulating hormone
Free T4
Metabolic panel
Renal function tests
Hepatic function tests
Serum vitamin B12 levels
Serum folate levels
Serological testing for syphilis
Serological testing for Lyme disease
Serological testing for HIV.
Results of haematological, serological, and biochemical tests are generally normal.
Brain imaging
The goals of imaging in those with suspected FTD are to:[103][104]
Assess for potentially treatable structural brain abnormalities (these might mimic a clinical presentation of FTD)
Identify features that support a diagnosis of FTD
Provide prognostic information and help initiate earlier treatment.
Magnetic resonance imaging (MRI) is the key imaging modality for diagnosing FTD; computed tomography (CT) should be ordered if a diagnosis of FTD is suspected but MRI is not available or is contraindicated.
MRI demonstrates classic atrophy patterns associated with FTD, which predominantly affect the frontal and temporal lobes.[104]
CT can be used to exclude structural abnormalities such as masses or subdural haematoma that may present with frontal lobe dysfunction, but it may also show atrophy indicative of FTD.[104]
Fluorodeoxyglucose (FDG)-positron emission tomography (PET)/CT can help differentiate FTD from Alzheimer’s disease and dementia with Lewy bodies, showing hypometabolism mainly in the frontal and temporal lobes, often with left-right asymmetry.[103][104][105] It is most helpful when combined with MRI.[104]
A negative amyloid PET/CT can help rule out Alzheimer’s disease, but may be positive in a small proportion of patients with FTD, possibly indicating a mixed pathology.[104] Brain perfusion single photon emission computed tomography (SPECT) may detect hypoperfusion but is generally considered less helpful than FDG-PET/CT in the initial imaging of suspected FTD.[104]
Subtypes of FTD may be determined by observing specific patterns of atrophy and hypometabolism:[104][106][107][108][109][110][111][112]
bvFTD is typically associated with atrophy on structural MRI and hypometabolism on FDG-PET in the prefrontal cortex and anterior temporal lobes, with relative sparing of more posterior regions of the brain, such as the occipital lobe. Disruption in the structural connectivity of the brain, measured using diffusion tensor imaging (DTI), is also a feature.
Semantic dementia is associated with characteristic patterns of atrophy on structural MRI, and hypometabolism in the anterior temporal lobes is evident on FDG-PET.
Primary progressive aphasia (PPA) is associated with relatively focal patterns of atrophy on MRI, and hypometabolism on FDG-PET in the left posterior frontal lobe.
Logopenic variant PPA shows atrophy on MRI and hypometabolism on FDG-PET in the lateral temporoparietal and medial parietal lobes (left greater than right), and left frontal lobe.
Progressive supranuclear palsy is associated with atrophy of the midbrain on MRI (which may manifest as the hummingbird, Mickey mouse, or morning glory signs) and hypometabolism of midbrain and frontal lobe on FDG-PET.
In corticobasal degeneration, MRI shows asymmetric cortical atrophy involving the posterior frontal and parietal regions, along with dilatation of the lateral ventricles. FDG-PET demonstrates hypometabolism in the superior parietal and superior frontal regions, and less frequently in the basal ganglia and thalamus.
In patients with bvFTD and amyotrophic lateral sclerosis MRI may show atrophy in premotor regions and precentral gyri.
Advanced MRI methods (e.g., voxel-based morphometry, resting-state functional MRI, DTI, and arterial spin labelling and anteroposterior index) may differentiate between FTD and Alzheimer's disease, with a reported sensitivity of 60% to 83% and specificity of 63% to 93%.[103][113]
Studies of the fine structure of the adult neuronal networks and specific sub-regional network 'hubs' are likely to provide normative benchmarks in the differential diagnosis of common dementias including FTD.[114] Broadly, in FTD, white matter disruption is more severe than grey matter damage.[115]
[Figure caption and citation for the preceding image starts]: Neuroimaging patterns associated with behavioural variant FTD (bvFTD) and nonfluent variant primary progressive aphasia (nfvPPA). Structural MRI and FDG-PET demonstrating the variability in patterns of atrophy and hypometabolism in FTD. In the case of bvFTD, significant bilateral frontal lobe atrophy and hypometabolism is seen. In the case of nfvPPA, atrophy and hypometabolism is lateralised and is greatly impacting the left frontal lobe more so than the right.Peet BT et al. Neurotherapeutics 2021 Apr; 18 (2): 728-52; used with permission [Citation ends].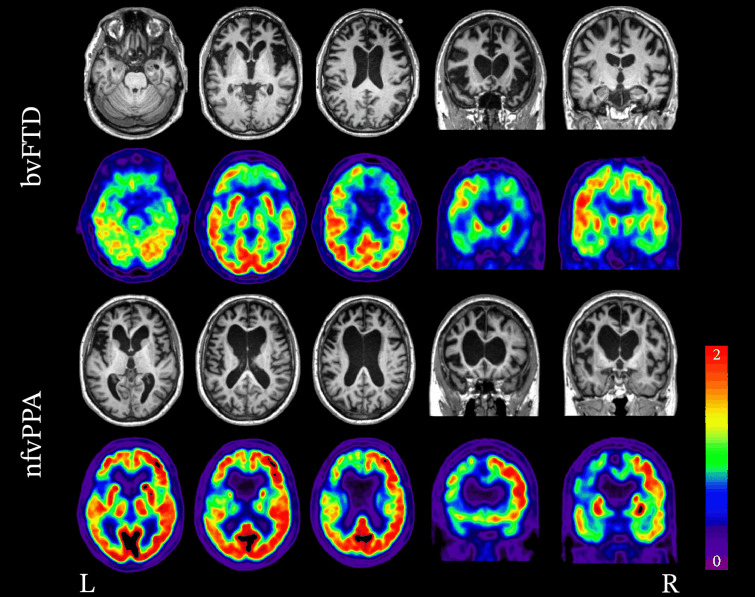
[Figure caption and citation for the preceding image starts]: Coronal T1-weighted MRI (a, b) and fused FDG-PET MRI (c) in a patient with semantic dementiaBhogal P et al. Eur Radiol 23, 3405-17 (2013); used with permission [Citation ends].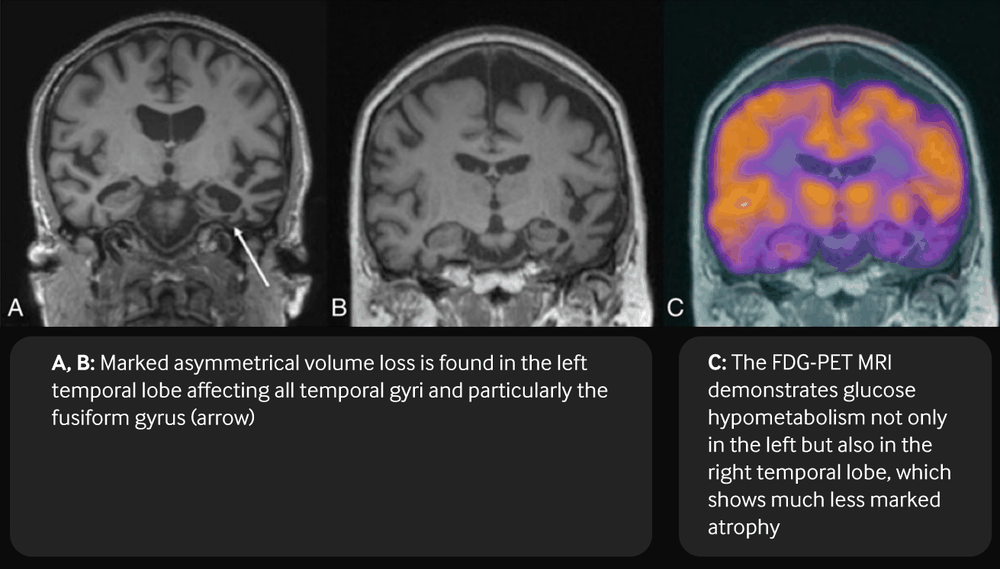
Genetic testing
Genetic tests to identify a known relevant mutation are not generally available in routine clinical genetic services. The clinical context is rarely straightforward; a clinical diagnosis of the subtype of dementia is often provisional, and neuropathology is lacking for affected family members. If genetic testing is undertaken, the clinical genetic team requires sufficient laboratory resources to distinguish between subtypes of dementia.[116]
Family history of FTD is a good indicator of whether genetic testing is appropriate. Mutations in the GRN and MAPT genes are present almost exclusively in patients with a strong family history, whereas C9orf72 expansion can occur in apparently sporadic disease.[117] Based on clinical experience and available evidence, genetic testing is desirable for all patients with probable or possible bvFTD, and in patients with suspected bvFTD with strong psychiatric features and at least one affected family member. Screening for C9orf72 mutations should take place for all patients with suspected FTD with prominent psychiatric symptoms or family history of late-onset primary psychiatric disorder (even if full diagnostic criteria are not met).[28] Genetic testing (for MAPT, GRN, and C9orf72) should also be performed if the results could contribute to decisions about pregnancy.
Genetic testing should be considered within the context of a clinical genetic service with the capacity to provide educational and psychological support for affected families. Pre- and post-test counselling is recommended. Genetic counselling assesses family history, and advises on genetic testing and its impact. It helps individuals and families understand the potential risks and implications of FTD, particularly familial types, and facilitates decisions about genetic testing and management of the condition. Following testing, counselling helps with interpreting results, and provides support and information about inheritance patterns and potential risks for other family members.[118][119]
Other tests
An electroencephalogram (EEG) or electromyogram (EMG) may be indicated in specific circumstances to rule out other diagnoses.
EEG
EEG use in dementia is endorsed when there has been a history of seizures or altered consciousness, raising the possibility of epilepsy, encephalopathy, or atypical subacute course.[106][120] Patterns of lateralised or bilateral epileptiform abnormalities, or slow (sometimes triphasic and periodic) waves support a diagnosis of late-onset epilepsy, prion disease, or toxic, metabolic, septic, autoimmune, and anoxic encephalopathies.[106][121][122][123]
Although scant data exist, research to date supports a potential use for electroencephalography and magnetoencephalography in identifying FTD.[124] Results from one small study show electroencephalography integrated with machine learning to detect FTD with high accuracy; further analysis of this method in this setting is required to confirm validity.[125]
EMG
An EMG may be indicated if symptoms and signs suggestive of amyotrophic lateral sclerosis are noted. In lower motor neuron dysfunction, an EMG may show evidence of chronic neurogenic change (large motor unit potentials of increased duration and/or increased amplitude), and evidence of ongoing denervation, (fibrillation potentials or positive sharp waves, or fasciculation potentials).[126]
Brain tissue examination
Neuropathological examination is the standard for definitive diagnosis. Specimens may be obtained by brain biopsy, but this is generally not recommended. Therefore, pathological confirmation is typically obtained at the end of life, and is particularly valuable in the characterisation of familial dementia.
The neuropathological diagnosis is based on formal criteria.[127][128] On gross examination, regional atrophy predominantly affecting the frontal and/or temporal lobes is found. Molecular neuropathology allows most cases of FTD to be placed into one of three molecular subgroups: frontotemporal lobar degeneration with tau (30% to 50% of cases), TAR DNA-binding protein (TDP-43), or FET protein accumulation.[53]
Fluid biomarkers
There is a growing interest in exploring potential fluid biomarkers in cerebrospinal fluid (CSF), serum and plasma for diagnostic, prognostic, and staging purposes.
There is no specific single fluid biomarker that uniquely identifies FTD pathologies associated with sporadic cases. Various markers have been and continue to be studied. Using a combination of markers may potentially increase diagnostic accuracy.[129]
Compared with Alzheimer’s disease, FTD is characterised by higher levels of CSF Aß42, and lower t-tau/Aß42 and p-tau/Aß42 values. The ratio of p-tau/Aß42 appears to be the most sensitive and specific biomarker for discriminating FTD with primary language disturbances from Alzheimer’s disease.[130] It has also been shown that plasma p-tau-181 and p-tau-217 are raised in Alzheimer’s disease but not FTD, except for certain MAPT mutations.[131][132] In patients with TAR DNA-binding protein 43 (TDP-43) proteinopathy, there is a reduction in the ratio of p-tau181 to total tau.[133]
Use of this content is subject to our disclaimer