História e exame físico
Principais fatores diagnósticos
comuns
presença de fatores de risco
Os fatores de risco fortes incluem história familiar de puberdade tardia (pode ser específica para atraso constitucional ou hipogonadismo hipogonadotrófico orgânico), anormalidades congênitas da hipófise, mutações genéticas conhecidas, alterações cromossômicas, diagnóstico sindrômico, anosmia, transtornos alimentares, doença sistêmica crônica, desnutrição, exercício intenso e anormalidades gonadais congênitas e adquiridas.
meninos: testículos <4 mL
O tamanho dos testículos é documentado como a medida do eixo mais longo ou o volume dos testículos medido pelo orquidômetro de Prader.
[Figure caption and citation for the preceding image starts]: Orquidômetro de PraderCriado pelo BMJ Knowledge Centre [Citation ends]. [Figure caption and citation for the preceding image starts]: Método de comparação do tamanho testicular usando o orquidômetro de PraderDo acervo de Dra. A. Mehta [Citation ends].
[Figure caption and citation for the preceding image starts]: Método de comparação do tamanho testicular usando o orquidômetro de PraderDo acervo de Dra. A. Mehta [Citation ends].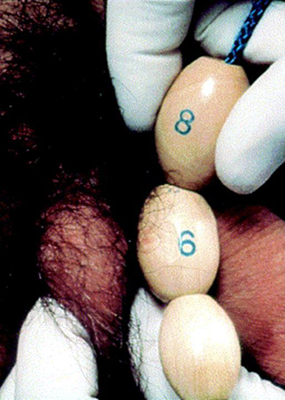 Um volume de 4 mL ou um comprimento longitudinal de 2.5 cm define o início da puberdade.[39]f
Um volume de 4 mL ou um comprimento longitudinal de 2.5 cm define o início da puberdade.[39]f
meninas: desenvolvimento das mamas ausente
O primeiro sinal visível de puberdade nas meninas é o desenvolvimento das mamas.[38]
ausência de pelos púbicos/axilares
Pelos púbicos e axilares, acne e odor corporal se desenvolvem como resultado de androgênios secretados pela glândula adrenal. O surgimento dos pelos axilares ocorre no meio da puberdade.
ausência de menarca >3 anos a partir do brotamento mamário
A menarca serve como evidência bem definida e documentada de puberdade em meninas.
Na maioria das meninas, ocorre tipicamente com o estágio 4 de desenvolvimento mamário de Tanner.
ausência de estirão de crescimento
Os hormônios sexuais estimulam diretamente a placa epifisária, resultando no estirão de crescimento. Há também um aumento na secreção do hormônio do crescimento (GH).[33] O estrogênio, seja do ovário ou aromatizado da testosterona testicular, é o fator que atua como mediador da resposta intensificada de GH durante a puberdade.[34]
O estirão de crescimento ocorre entre o meio e o fim da puberdade em meninos e no estágio 3 de desenvolvimento mamário em meninas.
Em média, contribui para 25 cm de altura nas mulheres e 30 cm nos homens.
anosmia
A síndrome de Kallman está associada ao hipogonadismo hipogonadotrófico orgânico e a nervos olfatórios hipoplásicos, resultando em anosmia.[21] Os outros sinais importantes associados ao hipogonadismo hipogonadotrófico orgânico (e conhecidos como "sinais de alerta" para a condição) incluem criptorquidia bilateral, micropênis (2 ou mais desvios padrão menor que o comprimento médio para a idade), defeitos da linha média, como fenda labial, fenda palatina e agenesia renal, bem como sincinesia (movimentos de espelho), a qual é patognomônica para esta condição.
Outros fatores diagnósticos
comuns
baixa estatura
O retardo constitucional e a síndrome de Turner estão associados à baixa estatura.
Incomuns
características dismórficas
Meninas com síndrome de Turner têm baixa estatura com algumas características dismórficas que incluem linha do cabelo posterior baixa, pescoço alado e orelhas proeminentes, com rotação posterior. A presença de sintomas pode ser mais sutil, e é necessário realizar investigações para descartar a síndrome de Turner em qualquer menina que apresente puberdade tardia.[40]
Os pacientes devem ser avaliados quanto à desproporção corporal. Meninos com síndrome de Klinefelter apresentam estatura alta, com maior crescimento dos membros inferiores. Além disso, eles têm testículos disgenéticos e atraso no desenvolvimento, e podem apresentar ginecomastia.[27]
Pode haver outros sintomas que sugiram um diagnóstico de síndrome (por exemplo, Prader-Willi, Bardet-Biedl, CHARGE [caracterizada por coloboma, atresia das cóanas e canais semicirculares anormais] ou displasia septo-ótica).
Fatores de risco
Fortes
história familiar de puberdade tardia
O atraso constitucional da puberdade, a causa mais comum de puberdade tardia em homens e mulheres, agrupa-se em famílias e exibe mais comumente um padrão de herança autossômico dominante.[1][19][20] Entre 50% e 75% dos indivíduos com atraso constitucional têm história familiar de puberdade tardia.[1]
Há uma semelhança na idade da puberdade entre meninas e suas mães, principalmente na idade da menarca.
anormalidades estruturais congênitas da hipófise
Pode decorrer de mutações genéticas ou anormalidades estruturais do eixo hipotálamo-hipófise associadas a defeitos na linha média do prosencéfalo (por exemplo, displasia septo-ótica e holoprosencefalia).[22] A deficiência de gonadotropina resultante pode ser isolada ou combinada com deficiências de outros hormônios da hipófise.
mutações genéticas
Alguns genes estão associados à patogênese do hipogonadismo hipogonadotrófico, como o ANOS1, FGFR1, KISS1R, KISS-1, GNRHR, PROK2, PROKR2, NSMF, NROB1, LH e a subunidade beta do FSH, leptina, receptor de leptina e pro-hormônio convertase 1 (PC1).[24]
alterações cromossômicas
A síndrome de Klinefelter (XXY), a síndrome de Turner (45X), a disgenesia gonadal XY e a disgenesia gonadal mista 45X/46XY estão associadas a gônadas disgenéticas e a hipogonadismo hipergonadotrófico.[27] Os homens com síndrome de Klinefelter comumente entram na puberdade, mas não conseguem evoluir completamente durante a puberdade, com aumento das concentrações de hormônio folículo-estimulante e queda das concentrações de testosterona a partir da metade da puberdade.
diagnóstico de síndrome
As síndromes Prader-Willi, Bardet-Biedl e CHARGE (caracterizada por coloboma, atresia das cóanas e canais semicirculares anormais) estão associadas a hipogonadismo hipogonadotrófico.
alimentação restritiva
A anorexia nervosa e padrões alimentares anormais podem resultar em hipogonadismo hipogonadotrófico.
doença sistêmica crônica
Doenças comuns, incluindo cardiopatia crônica, asma moderada a grave, fibrose cística, doença celíaca, doença inflamatória intestinal (doença de Crohn e colite ulcerativa), distúrbios inflamatórios (por exemplo, artrite idiopática juvenil), insuficiência renal crônica, qualquer neoplasia maligna crônica e diabetes mellitus mal controlado podem causar hipogonadismo hipogonadotrófico.
desnutrição
Pode resultar em hipogonadismo hipogonadotrófico funcional.
exercícios físicos intensos
O exercício físico intenso pode resultar em retardo no início da puberdade, retardo na menarca ou interrupção do desenvolvimento puberal.
anormalidades testiculares congênitas
O hipogonadismo hipergonadotrófico pode ser decorrente de anorquia ou regressão testicular nos homens. A criptorquidia bilateral grave (testículos que não desceram) também pode estar associada a anormalidades testiculares congênitas.
anormalidades gonadais adquiridas
O hipogonadismo hipergonadotrófico pode decorrer de torção ou tumor testicular.
A endocrinopatia autoimune pode resultar em dano gonadal e hipogonadismo hipergonadotrófico.[29]
cirurgia na hipófise
Pode resultar em hipogonadismo hipogonadotrófico (por exemplo, após remoção de um craniofaringioma ou adenoma).[25]
hipoplasia adrenal
Mutações do NROB1 causam hipogonadismo hipogonadotrófico e hipoplasia adrenal congênita, a qual pode levar a uma crise adrenal neonatal grave. As outras mutações implicadas na hipoplasia adrenal congênita incluem aquelas em STAR, CYP11A1 e CYP17A1.[35] A condição é herdada como um distúrbio ligado ao cromossomo X.[23]
Fracos
quimioterapia
Pode causar hipogonadismo hipergonadotrófico ou hipogonadotrófico, dependendo do agente quimioterápico usado. O risco da quimioterapia é maior com os agentes alquilantes.[36]
radioterapia
Pode resultar em hipogonadismo hipogonadotrófico com irradiação no cérebro/hipófise ou hipogonadismo hipergonadotrófico após irradiação na pelve.[30]
histiocitose
Pode resultar em hipogonadismo hipogonadotrófico permanente.
doença falciforme
Pode resultar em hipogonadismo hipogonadotrófico permanente.
sobrecarga de ferro (associada a transfusão)
Pode resultar em hipogonadismo hipogonadotrófico permanente.[26]
O uso deste conteúdo está sujeito ao nosso aviso legal