Etiologia
A pancitopenia pode ocorrer devido à redução da produção de células ou falência da medula óssea, distúrbios clonais da hematopoiese, sequestro ou destruição não mediados imunologicamente elevados, ou uma destruição mediada imunologicamente das células sanguíneas.[Figure caption and citation for the preceding image starts]: Tabela de etiologias para pancitopenia (LES: lúpus eritematoso sistêmico, CMV: citomegalovírus, EBV: vírus Epstein-Barr, DECH: doença do enxerto contra o hospedeiro)Do acervo de Jeff K. Davies [Citation ends].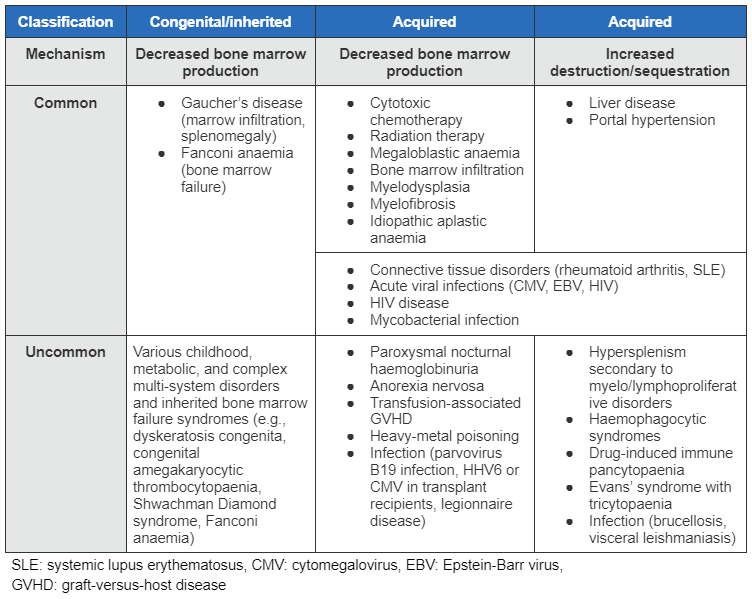
Produção reduzida na medula óssea
A medula óssea é o local onde são produzidos eritrócitos, leucócitos e megacariócitos, de onde vêm as plaquetas. Quando as células são criadas, elas são liberadas na circulação periférica. Esse processo requer uma atividade adequada das células-tronco hematopoiéticas e um ambiente estromal da medula óssea funcionante. A alta taxa de proliferação da medula requer um estado nutricional adequado, particularmente de vitamina B12 e ácido fólico, além de quantidades vestigiais de outros elementos.
Quimioterapia
As causas mais comuns de pancitopenia transitória em todas as faixas etárias são a quimioterapia citotóxica e a radioterapia.
A pancitopenia relacionada à quimioterapia raramente representa um dilema diagnóstico e geralmente se resolve em 1 a 2 semanas (a menos que haja uma síndrome de falência medular hereditária não reconhecida). Alguns indivíduos podem ter defeitos proliferativos conhecidos ou não, ou uma farmacogenética específica, o que pode predispô-los a uma pancitopenia mais grave e de maior duração. Alguns esquemas terapêuticos estão associados a períodos significativamente mais longos de pancitopenia. A pancitopenia relacionada à quimioterapia que dura mais do que o esperado deve ser investigada.
Anemia megaloblástica
Embora a maioria dos casos de anemia megaloblástica cause uma anemia macrocítica sem leucopenia ou trombocitopenia, uma anemia megaloblástica grave pode resultar em pancitopenia. A anemia megaloblástica resulta mais comumente da deficiência de vitamina B12 (por exemplo, anemia perniciosa, uma condição autoimune na qual autoanticorpos interferem na função do fator intrínseco, necessário para a absorção da vitamina B12 no trato gastrointestinal). Menos comum, a deficiência de vitamina B12 pode decorrer de dieta alimentar deficiente (em veganos) ou má absorção intestinal.
A deficiência de ácido fólico, quase sempre de origem alimentar, também resulta em anemia megaloblástica.
Infiltração da medula óssea
A infiltração da medula óssea é uma causa comum de pancitopenia e, geralmente, resulta de doença maligna. Em geral, o infiltrado é celular e pode ser de origem hematológica (por exemplo, leucemia linfoide e mieloide aguda, mieloma, linfoma não Hodgkin, leucemia de células pilosas, leucemia linfocítica crônica e mielofibrose) ou neoplasias de origem não hematológica (por exemplo, mama, pulmão, rim, próstata e tireoide).
Em crianças, a pancitopenia pode ser causada por neuroblastoma, rabdomiossarcoma, sarcoma de Ewing e retinoblastoma.[1]
Doenças do armazenamento lisossomal
Doenças de armazenamento lisossomal (por exemplo, doença de Gaucher) podem infiltrar a medula, resultando em pancitopenia. O infiltrado pode ser majoritariamente uma fibrose reticulínica, que também está associada a condições malignas.
Pacientes com doença de Gaucher podem apresentar esplenomegalia maciça e hiperesplenismo funcional, em associação com infiltração na medula óssea.[2]
Outras causas
Causas mais raras de pancitopenia decorrentes de uma menor produção de células sanguíneas pela medula óssea incluem anorexia nervosa, doença do enxerto contra o hospedeiro associada a transfusões em pacientes imunossuprimidos e envenenamento por metais pesados (por exemplo, arsênico).[3]
Infecções, como as causadas pelo HIV, foram associadas a pancitopenia secundária à subprodução medular (que frequentemente afeta a produção de eritrócitos), bem como a infecção pelo parvovírus em indivíduos com fatores predisponentes específicos (anemia hemolítica; principalmente anemia falciforme e esferocitose hereditária).[4]
Distúrbios clonais de hematopoiese
A síndrome mielodisplásica (SMD) é um distúrbio clonal comum adquirido de células hematopoiéticas, caracterizado por hematopoiese ineficaz e displásica e uma propensão à evolução para leucemia mieloide aguda. Os adultos geralmente estão na casa dos 70 anos no momento do diagnóstico.[5][6] Em crianças, o termo citopenia refratária da infância (CRI) costuma ser usado. Crianças com SMD/CRI geralmente têm uma predisposição de linha germinativa subjacente (síndrome de falência medular hereditária, RUNX1, ANKRD26, ETV6, GATA2 e SAMD9/SAMDL9) para SMD em aproximadamente 30% a 40% dos casos.[7]
A medula óssea pode ser hipercelular ou hipocelular. Em ambos os casos, é comum vir acompanhada de pancitopenia do sangue periférico. Em associação com a produção reduzida ou inadequada de células sanguíneas na medula, algumas vezes há um mecanismo imunologicamente mediado que contribui para a pancitopenia do sangue periférico na SMD.
A hemoglobinúria paroxística noturna (HPN) é um distúrbio clonal adquirido raro (taxa de incidência de 5.7 por 1,000,000 pessoas-ano) das células hematopoiéticas, causado por mutação somática do gene do fosfatidilinositol glicano A ligado ao cromossomo X, que resulta em expressão deficiente de proteínas ancoradas por glicosilfosfatidilinositol.[8][9] A HPN é clinicamente caracterizada por trombose e hemólise intravascular, e a evolução com pancitopenia é comum (ela provavelmente surge da combinação de produção reduzida na medula óssea, secundária a defeitos adquiridos em células-tronco hematopoiéticas, com a destruição de células). Há uma sobreposição de características clínicas e laboratoriais entre pacientes com HPN e anemia aplásica idiopática (AAI) e até mesmo SMD.[Figure caption and citation for the preceding image starts]: Síndrome mielodisplásica hipoplásica com normoblastos displásicosMorris Edelman, MD e Peihong Hsu, MD [Citation ends].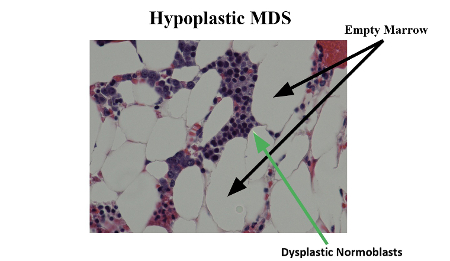
Falência medular
As síndromes de falências medulares hereditárias e congênitas se manifestam mais frequentemente na infância, embora diagnósticos em idade adulta estejam aumentando em função da conscientização e dos exames mais detalhados.
Anemia de Fanconi: predominantemente uma doença autossômica recessiva (uma rara herança dominante ligada ao cromossomo X foi descrita) na qual mais de 20 proteínas disfuncionais resultam em hematopoiese reduzida e falência da medula óssea.[10] A anemia de Fanconi apresenta várias características, como baixa estatura, hiperpigmentação, anormalidades do esqueleto, maior incidência de tumores sólidos e leucemia e uma sensibilidade celular aumentada a agentes que danificam o ácido desoxirribonucleico (DNA).[11][12][13]
Disceratose congênita (DC): hereditária como doença autossômica dominante ou doença autossômica recessiva ligada ao cromossomo X, é decorrente de lesões genéticas que comprometem a integridade de telômeros, resultando em perda da autorrenovação e regeneração das células.[14] Mutações em 14 genes associados à biologia de telômeros podem ser identificadas na maioria dos pacientes com características clínicas de DC clássica.[15][16][17] A DC clássica é definida por distrofia das unhas, leucoplasia das mucosas e alterações na pigmentação da pele, todas com variações de gravidade desde virtualmente não existente a grave.[13] Outras anormalidades incluem falência da medula óssea, calvície e cabelos grisalhos prematuros, estenoses uretrais, produção excessiva de lágrimas e fibrose pulmonar.[18]
Anemia aplásica idiopática (adquirida) (AAI): é uma doença adquirida rara (2-6 casos por milhão de habitantes). O diagnóstico de AAI requer a presença de pancitopenia em conjunto com celularidade reduzida da medula óssea sem infiltração ou fibrose.[19] A AAI é, portanto, diagnosticada por exclusão e deve ser cuidadosamente diferenciada da SMD hipocelular e de outras síndromes de falência medular congênitas e hereditárias.[20] Alguns pacientes têm história prévia de infecção viral, hepatite ou exposição a drogas. A AAI grave (em que a neutropenia e trombocitopenia são mais profundas) é uma condição que representa risco de vida.
Outras citopenias de célula única hereditárias raras: anemia de Diamond-Blackfan (ADB), síndrome de Shwachman-Diamond (SSD) e trombocitopenia amegacariocítica (AMT), podem evoluir para pancitopenia.[13]
Usando-se o sequenciamento completo do exoma e do genoma, mutações genéticas raramente relatadas em falências hereditárias da medula óssea foram identificadas em pacientes com falência medular de origem supostamente hereditária, mas sem um diagnóstico definido.[21][Figure caption and citation for the preceding image starts]: Anemia aplásica: a medula óssea normocelular é mostrada à esquerda; e a medula vazia, típica da anemia aplásica congênita ou adquirida, é mostrada à direitaMorris Edelman, MD e Peihong Hsu, MD [Citation ends].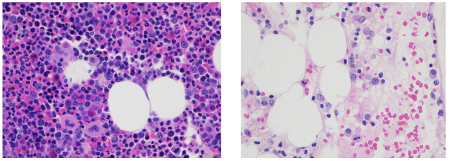
Destruição ou sequestro elevados
A maioria dos casos de pancitopenia acompanhados por produção adequada de células sanguíneas pela medula óssea são resultantes de maior sequestro de células sanguíneas no baço. Doenças que resultam em pancitopenia por hiperesplenismo funcional incluem:
Doença hepática (com hipertensão portal associada) causada por cirrose hepática alcoólica, infecção crônica por hepatite B ou C, hepatite autoimune, ou hipertensão portal idiopática.
Doenças mieloproliferativas (por exemplo, a leucemia mieloide crônica pode apresentar esplenomegalia maciça, resultando em pancitopenia apesar de uma produção adequada de células sanguíneas na medula óssea). Essas doenças raramente ocorrem em crianças.
Infecções agudas e crônicas que resultam em hiperesplenismo (por exemplo, brucelose e leishmaniose visceral). Considerações sobre exposição e histórico de viagens recentes são relevantes.
Síndromes hemofagocíticas (um grupo heterogêneo de distúrbios caracterizado por maior atividade de histiócitos ou macrófagos na medula óssea e em outros órgãos). Hepatomegalia e esplenomegalia são características clínicas comuns. Síndromes hemofagocíticas podem ser classificadas como primárias (nas quais a síndrome hemofagocítica domina as características clínicas da doença, como ocorre na linfo-histiocitose hemofagocítica primária [LHHp]), que, geralmente, têm origem genética, ou podem ser reativas a doenças sistêmicas com uma gama de outras características clínicas (por exemplo, distúrbios autoimunes, linfoma de células T, muitas vezes denominado síndrome de ativação de macrófagos). Em alguns casos, pode ser difícil diferenciar entre essas distinções, mas o teste genético está disponível para muitos dos genes envolvidos na LHHp.[22]
Destruição de células sanguíneas mediada imunologicamente
A pancitopenia imune induzida por medicamento ocorre quando há a geração de anticorpos com reatividade cruzada para medicamentos e células hematopoiéticas. Isso geralmente está associado a quinina, sulfonamidas, metotrexato e rifampicina.[23]
A pancitopenia imune pode ser observada em até 20% dos pacientes com síndrome de Evans (classicamente, a combinação de trombocitopenia autoimune e anemia hemolítica), mais comum em crianças que em adultos.[24] Um número significativo de pessoas com síndrome de Evans apresenta síndrome linfoproliferativa autoimune subjacente.
A síndrome linfoproliferativa autoimune (SLPA) é uma doença hereditária resultante de mutações que inibem a apoptose na regulação da resposta imune. Há relatos de casos leves, o que sugere que a incidência é significativamente subestimada. A SLPA é caracterizada por uma linfoproliferação geralmente benigna (linfadenopatia e esplenomegalia) e autoimunidade, na maioria das vezes direcionada às células da linhagem mieloide (eritrócitos, granulócitos e plaquetas), embora outros alvos sejam menos comumente envolvidos (por exemplo, hepatite autoimune).[25]
Pancitopenia por combinação
Várias condições associadas à pancitopenia resultam de uma combinação da redução de produção da medula óssea e aumento da destruição ou sequestro de células sanguíneas. Eles incluem:
Doenças do tecido conjuntivo (mais comumente artrite reumatoide e lúpus eritematoso sistêmico)
Infecção aguda por citomegalovírus
Infecção micobacteriana
Mononucleose infecciosa
vírus da imunodeficiência humana (HIV)
Síndrome de Felty (artrite reumatoide, esplenomegalia e neutropenia).
O uso deste conteúdo está sujeito ao nosso aviso legal