Aetiology
Human haemoglobin consists of a tetramer of two pairs of globin chains: one pair of alpha-like chains and one pair of non-alpha chains, each of which contains a haem group, and has the essential function of carrying oxygen around the body from the lungs. The alpha-globin gene cluster on each chromosome 16 contains one embryonic zeta gene and two co-expressed alpha-globin genes, alpha-2 and alpha-1. Normally, the alpha-2 gene, located up-stream of the alpha-1 gene, encodes 2- to 3-fold more protein than the alpha-1 gene.[24]
There are two major varieties of alpha-thalassaemia: alpha(0) thalassaemia (--/), in which both alpha-globin genes on the same chromosome are deleted, and alpha(+) thalassaemia (-alpha/), in which one of the two alpha-globin genes on the same chromosome is deleted or mutated. Non-deletional variants in general have a more severe phenotype than the more common deletional variants.[5] This seems to be primarily due to the mutated alpha-2 gene interfering with the ability of the alpha-1 gene to increase output.[1]
Clinical manifestations are also influenced by whether or not concomitant defects in beta-globin chain synthesis are present. Of note, simple carriers of multiplicated alpha genes (that is, extra copies of alpha-globin genes) are asymptomatic.
Pathophysiology
Alpha-thalassaemia is characterised by decreased or absent production of at least one of the four alpha-globin genes, and the clinical phenotype corresponds to the degree of impairment in alpha-globin chain synthesis, which likewise correlates largely with the genotype. The pathophysiology thus depends in small part on the degree of impairment in Hb assembly, but in large part is due to the accumulation of excess unmatched beta-globin chains, which aggregate, causing oxidant and mechanical damage to the affected red cells and leading to their premature destruction.
The clinical features of alpha-thalassaemia are predominantly those associated with anaemia and increased haemolysis.
Reduced alpha-globin chain synthesis and decreased Hb A (alpha2beta2) leads to the characteristic microcytosis and hypochromia seen with the disease. In addition, the decreased alpha-globin production leads to an excess of free non-alpha chains, and formation of gamma4 (Hb Bart) and beta4 (Hb H) tetramers. Hb Bart and Hb H have a high oxygen affinity, exhibit neither haem-haem interaction nor Bohr effect, and thus are non-functional as oxygen carriers.[25] The clinical manifestations depend on the degree of impairment in alpha-globin chain synthesis, and thus will correlate with the genetic defect.
Haemolysis, predominantly extravascular destruction, is seen in haemoglobin H (Hb H) and Hb H/Constant Spring but also in homozygous Hb Constant Spring disease (alpha(CS)alpha/alpha(CS)alpha).[6][26][27][28] Hb H and Hb H/Constant Spring red cells are abnormally rigid, possibly secondary to increased globin/red cell membrane associations impairing the red cell's ability to navigate the circulation.[29][30] Hb H also has increased susceptibility to oxidant injury, leading to precipitation of beta aggregates.[26] Oxidation of haem leads to the formation of haemichromes, which may in turn damage the red cell membrane.[31][32] Damage to the red cell membrane may lead to movement of phosphatidylserine from the inner to the outer red cell membrane leaflet and premature clearance of these cells.[33] Haemolysis in individuals with Hb H disease can be exacerbated by ingestion of oxidant drugs, infection, and fever. Haemolysis can also be exacerbated by co-existent G6PD deficiency, particularly upon exposure to oxidant stress. Patients with homozygous Hb Constant Spring and Hb H/Constant Spring also have a significant component of ineffective erythropoiesis contributing to the anaemia.[28]
Classification
Types and variants
There are two alpha-globin genes on each chromosome 16, labelled alpha-2 and alpha-1.[1] Thus, each person normally has a total of four functioning alpha-globin genes. Alpha-thalassaemia is characterised by reduced output of alpha-globin chains secondary to deletion and/or mutation of ≥1 of the 4 alpha-globin genes.
Classically, alpha-thalassaemia is sub-divided into two major types: alpha(0) thalassaemia (--/), in which both alpha-globin genes on the same chromosome are deleted, and alpha(+) thalassaemia (-alpha/), in which only one of the two alpha-globin genes on the chromosome is deleted or mutated. Large deletions involving the alpha-globin genes may also include the HS-40 regulatory element or, rarely, alpha(0)-thalassaemia may be caused by deletions of HS-40 that leave the alpha-globin genes intact.[3][4]
Deletions leading to alpha-thalassaemia are more common than non-deletional variants. The seven most common alpha-thalassaemia deletions are -alpha(3.7) (common in African-Americans), -alpha(4.2), (--FIL), (--THAI), (--MED), -(alpha)(20.5), and (--SEA). In general, non-deletional variants leading to alpha-thalassaemia have a more severe phenotype than deletional variants.[5] This appears to be primarily due to a mutated alpha-2 gene interfering with the ability of the normal alpha-1 gene to increase output.[1] In addition, large deletions or those affecting the regulatory element may also lead to a more severe clinical phenotype.[3]
Genotypic and phenotypic classification
The clinical phenotype corresponds with the genetic defect and the degree of impairment in alpha-globin chain synthesis. The spectrum of disease ranges from the clinically unremarkable silent carrier state to alpha-thalassaemia major, almost uniformly fatal in utero without intervention. Haemoglobin H (Hb H) disease most often leads to an intermediate phenotype, with a moderate to severe anaemia and haemolysis.
There are at least four different and distinct alpha-thalassaemia sub-types.[1]
Alpha-thalassaemia silent carrier
Silent carrier state occurs when only 1 of the 4 alpha-globin genes is affected.
Patients are likely to be asymptomatic and haematologically normal.
Alpha-thalassaemia silent carrier is also known as alpha-thalassaemia silent trait, alpha-thalassaemia-2 trait, heterozygosity for alpha(+) thalassaemia, and alpha-thalassaemia minor.
Alpha-thalassaemia trait
Alpha-thalassaemia trait occurs when 2 of the 4 alpha-globin genes are affected: for example, either heterozygosity for alpha(0) thalassaemia (that is, 2 alpha-globin genes on the same chromosome, in cis, are deleted) or homozygosity for alpha(+) thalassaemia (that is, 1 alpha-globin gene on each chromosome, in trans, is deleted or mutated).
Patients with alpha-thalassaemia trait may have a mild asymptomatic anaemia, and physicians often mistakenly diagnose these patients as having iron deficiency anaemia.
Importantly, patients who are homozygous for non-deletional alpha(+) thalassaemia may have more severe manifestations of the disease. This is the case with Haemoglobin Constant Spring, which is caused by a mutation in the alpha-2 globin gene. Patients who are homozygous for this mutation (that is, both alpha-2 globin genes are affected) have a more serious clinical phenotype than those who are homozygous for deletional alpha(+) thalassaemia. They have a mild anaemia with a normal mean corpuscular volume (MCV) and slightly low mean corpuscular haemoglobin (MCH), and frequently have jaundice and splenomegaly.[6]
Haemoglobin H (Hb H) disease
Hb H disease typically affects 3 alpha-globin genes (beta4 tetramers). [Figure caption and citation for the preceding image starts]: Haemoglobin H diseaseFrom the collection of Elizabeth A. Price and Stanley L. Schrier, Stanford University [Citation ends].
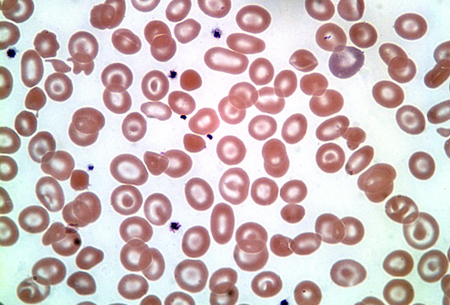 It is most commonly caused by deletion of 3 alpha-globin genes, but can also be caused by deletion of 2 alpha-globin genes with an inactivating point mutation of a third gene.
It is most commonly caused by deletion of 3 alpha-globin genes, but can also be caused by deletion of 2 alpha-globin genes with an inactivating point mutation of a third gene. Atypical Hb H disease (or even hydrops fetalis) can also be caused by homozygous non-deletion mutations such as polyadenylation signal mutations in the alpha-2 globin gene.[7]
Hb H may be detected in the peripheral blood on routine Hb electrophoresis. Hb H inclusion bodies may also be demonstrated on supravital staining.
Alpha-thalassaemia major
Typically caused when all 4 alpha-globin genes are deleted.
It is also known as haemoglobin Bart hydrops fetalis syndrome or homozygous alpha(0) thalassaemia.
Use of this content is subject to our disclaimer