Details
Congenital heart disease (CHD) is the most common birth defect, although still relatively rare. Centers for Disease Control and Prevention: facts about congenital heart defects Opens in new window Screening for fetal CHD includes ultrasonography in the second trimester of pregnancy and postnatal clinical examination; however, detection rates are low.[1][2][3][4]
Early recognition of CHD is important because clinical presentation and deterioration may be sudden and some treatable defects may even cause death before diagnosis.[1]
The surgical and medical treatment of CHD has markedly improved over the last 50 years. The first successful open heart surgeries for intracardiac defects were carried out in the US in the 1950s.[5][6][7] It was the introduction of machines that perfused the vital organs while a surgeon carefully repaired a non-beating heart that revolutionised the field of corrective surgery. Survival well into adulthood is now expected for most babies born with CHD.[8]
Worldwide, the prevalence of congenital heart disease (CHD) is estimated to be 0.8% to 1.2% of live births.[9] In high-income North America, including the US, the birth prevalence of CHD is estimated to be 12.3 per 1000 (95% CI, 10.9 to 13.8). An estimated 1% or a minimum of 40,000 infants per year are expected to be affected by CHD each year in the US. Around, 25%, or 2.4 per 1000 live births, will undergo invasive treatment in the first year of life. The most common type of congenital heart defect is bicuspid aortic valve, which occurs in 13.7 of every 1000 people.[10]
Chromosomal abnormalities (e.g., DiGeorge syndrome, Down's syndrome, or Turner's syndrome) are found in 8% to 10% of individuals with CHDs and single-nucleotide or pathogenic copy number variants are found in 5% to 15%.[10]
Recent reports show a significant decrease in mortality among patients with CHDs. In 2020, there were 2817 deaths related to CHD in the US, an 11.9% decrease from 2010, and the age-adjusted death rate was 0.9 deaths per 100,000 people, an 18.2% decrease from 2010.[10] Due to the improved survival, there is now an aging population of adults with CHD who present with complications related to acquired cardiovascular disease (e.g., thromboembolic events and arrhythmias).[8][11][12] The increased risk of morbidity and mortality from premature atherosclerotic cardiovascular disease (ASCVD) in this group, poses an increasing challenge and highlights the need for long-term monitoring of patients and awareness of their ASCVD risk factors.[8]
Left-to-right shunts
Lesions that allow blood to shunt from the left side to the right side of the heart. They are associated with varying degrees of increased pulmonary blood flow and are typically acyanotic. In some defects, the site of the shunt may not be within the heart itself.
Cyanosis occurs only if the lesions are large and not repaired in childhood, and if the patient develops pulmonary vascular obstructive disease (Eisenmenger's physiology). Echocardiography is the primary imaging modality, and, in the current era, the role of cardiac catheterisation is primarily for intervention.[13]
Examples include:
Ventricular septal defect (VSD)
Atrial septal defect (ASD)
Atrioventricular septal defect (AVSD)
Patent ductus arteriosus (PDA)
Partial anomalous pulmonary venous connection (PAPVC).
Right-to-left shunts
Lesions that result in de-oxygenated blood reaching the aorta and are associated with an increased or decreased pulmonary blood flow.
Examples include:
Tetralogy of Fallot (TOF)
Pulmonary valve atresia with or without a VSD
d-Transposition of the great arteries (d-TGA)
Truncus arteriosus
Ebstein's anomaly
Total anomalous pulmonary venous connection (TAPVC)
Hypoplastic left heart syndrome (HLHS).
Obstructive valvular and non-valvular lesions
Left ventricular outflow tract (LVOT) obstruction
Coarctation of the aorta
Pulmonary valve stenosis (PS)
Aortic valve stenosis (AS)
Congenital heart defects can also be classified using the anatomical/physiological (AP) classification introduced by the American Heart Association/American College of Cardiology (AHA/ACC) task force in their 2018 guidelines.[14]
In the AP system, a patient is classified with both an ‘A’ class and ‘P’ class based on the 'highest' relevant anatomical or physiological feature, and patients may move from one AP classification to another over time. If clinical status worsens, the classification will change to a higher severity group, but improvement in status; for example, after an intervention such as valve replacement or control of arrhythmia, can result in change to a lower severity classification.
In the 2020 guidelines for the management of adults with congenital heart defects published by the European Society of Cardiology (ESC), only the anatomical classification is used, without the physiological component.[15] There are differences in the severity rating across both classification systems; for example, congenital aortic valve stenosis and congenital mitral valve disease are considered mild defects in the ESC guidelines, whereas the AHA/ACC guidelines grade them as moderate.
Screening for fetal congenital heart disease (CHD) includes ultrasonography in the second trimester of pregnancy and postnatal clinical examination; however, detection rates are low.[1][2][3][4] Fetal echocardiography is becoming the definitive test for diagnosis. The American Society of Echocardiography has produced recommendations for when to refer for fetal imaging, although they acknowledge there are local differences in referral indications depending on the population being screened.[16]
In the US, the Centers for Disease Control and Prevention (CDC) recommends routine newborn screening for CHD, combining pulse oximetry with a physical examination, at age 24 hours or as late as possible before early discharge.[17] The American Academy of Pediatrics (AAP) endorses an algorithm for newborns using pulse oximetry, citing that screening using pulse oximetry has markedly reduced early infant mortality and emergency hospitalisations attributable to critical CHD.
The algorithm is simplified as follows compared to previous iterations:[18]
A passing oxygen saturation requires ≥95% in both pre- and post-ductal measurements (previously ≥95% in either).
Only one retest is required for indeterminate results instead of two.
Pulse oximetry should be measured in the right hand and one foot; if pulse oximetry is <90% in either measurement then immediate clinical assessment should follow. If pulse oximetry is ≥95% at both sites and the saturation difference is ≤3% then routine care may be provided; otherwise, measurements should be retaken after one hour. If the retaken oxygen saturations are <95% at either site or the saturation difference is >3%, then immediate clinical assessment should follow. Screening on room air is recommended.
This algorithm identifies asymptomatic term infants who should be assessed clinically for a number of types of CHD as well as for hypoxaemic conditions other than CHD.
In the UK, screening for CHD using physical examination is routinely performed as part of the newborn and infant physical examination (NIPE) screening programme; additionally, units are increasingly adopting pulse oximetry screening as an adjunct to this.[19]
VSD is the most common form of CHD, accounting for 20% of all cases, excluding bicuspid aortic valve and mitral valve prolapse.[20] Subtypes based on location include: peri-membranous (in the area of the membranous septum); outlet (below 1 or both semilunar valves); inlet (inferior to the atrioventricular [AV] valves); and muscular (in the muscular or trabeculated portion of the ventricular septum).[21][Figure caption and citation for the preceding image starts]: Subtypes of ventricular septal defects: (A) outlet; (B) perimembranous; (C) inlet; (D) muscularMayo Clinic Foundation [Citation ends].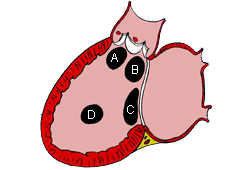
Peri-membranous defects typically occur as a solitary lesion and can sometimes close spontaneously by apposition of the septal leaflet of the tricuspid valve to the defect. Outlet defects may be large and associated with more complex forms of congenital heart disease such as tetralogy of Fallot. Both peri-membranous and outlet VSDs are in close proximity to the right cusp of the aortic valve. Because of the Venturi effect, these defects can cause prolapse of an aortic valve cusp, which results in both a restriction to flow through the VSD and regurgitation of the aortic valve.[22] Inlet defects do not close spontaneously and may be associated with AVSD and AV valve regurgitation. Muscular defects are the most common type of VSDs in newborns and the vast majority close spontaneously before 2 years of age.[Figure caption and citation for the preceding image starts]: Apical 4-chamber echocardiographic image of a muscular VSD (arrow). (RA) right atrium; (LA) left atrium; (RV) right ventricle; (LV) left ventricleImage courtesy of Patrick W. O'Leary, MD [Citation ends].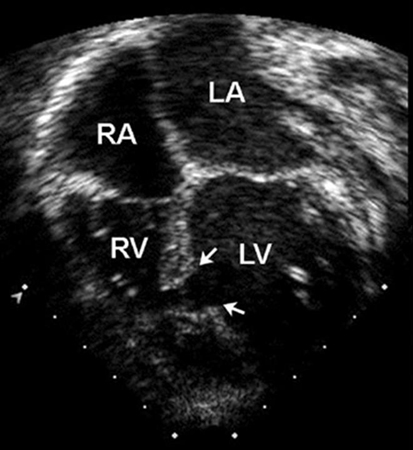
Children with a small VSD develop normally and are asymptomatic. With larger defects, the excess pulmonary blood flow that ensues after the age of 6-8 weeks when the normal decrease in the pulmonary blood flow occurs may lead to the developments of tachypnoea, tachycardia, pallor, poor feeding, and poor weight gain. Pulmonary vascular remodelling and obstructive disease usually do not develop in infants, but can develop by 2 years of age. If left untreated, most moderate to large VSD eventually lead to severe pulmonary hypertension and at some point reversal of the shunt occurs, leading to central cyanosis. This condition, called Eisenmenger's syndrome, does not typically occur until later in life. Once a patient develops Eisenmenger's physiology, closure of the VSD is contraindicated.[23][24]
Patients have a low-to-mid frequency holosystolic murmur, a prominent pre-cordial impulse, and, in patients with pulmonary hypertension, a loud and single second heart sound. If the left-to-right shunt is large, patients develop a mid-diastolic flow murmur ('rumble') across the mitral valve.
CXR and ECG can be normal in small VSDs. In larger defects, there is cardiac enlargement and increased pulmonary vascular markings on CXR, and ECG reveals left ventricle hypertrophy; right ventricle hypertrophy can occur with larger defects. Inlet-type VSDs are associated with left axis deviation on ECG. Echocardiography provides important information regarding the anatomy of the defect, the volume of the shunt, and right ventricular pressure.[25][26][Figure caption and citation for the preceding image starts]: CXR demonstrating pulmonary overcirculationMayo Clinic Foundation [Citation ends].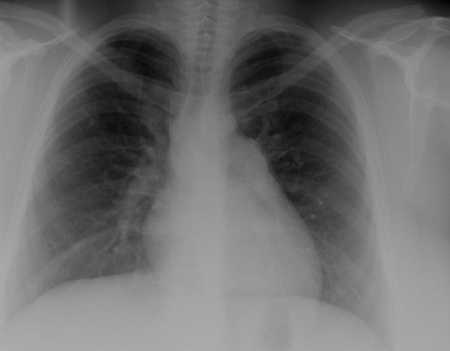
The goal of treatment is to ensure adequate somatic growth and prevent pulmonary vascular obstructive disease. In the absence of the development of pulmonary hypertension, VSDs typically lead to left ventricular (LV) enlargement, which serves as an indication for closure. Small VSDs with restrictive flow may be left untreated with need for continuous surveillance for the development of LV enlargement and endocarditis. Large VSDs and defects associated with aortic regurgitation due to aortic valve cusp prolapse or excess pulmonary blood flow may require diuretics and an increased caloric formula, followed by closure of the defect (either surgical closure or trans-catheter device closure). Those with increased pulmonary blood flow and faltering growth are closed surgically between 1 and 4 months of age; most large defects are typically closed by 6-9 months of age. For patients with Eisenmenger's syndrome, pulmonary vasodilator therapy is the treatment of choice, and women of childbearing age should be informed that pregnancy is not advised due to high maternal mortality and fetal complications.[23][24]
For more information see Ventricular septal defects.
Interatrial communications include atrial septal defects (ASDs) and other pre-tricuspid shunts (e.g., sinus venosus defect and unroofed coronary sinus). Pre-tricuspid shunts are defined as the presence of a left-to-right shunt that occur proximal to the right atrio-ventricular valve. As compared to post-tricuspid shunts (such as ventricular septal defect and patent ductus arteriosus), these defects are characterised by low-pressure shunting and varying degree of volume shunting. Pre-tricuspid shunts represent 6% to 10% of all CHDs and have a female-to-male predominance of 2:1.[27] The main types consist of ostium secundum atrial septal defect (60% to 70% of all pre-tricuspid shunts), which occur in the area of the fossa ovalis; ostium primum defect, a form of atrioventricular septal defect (AVSD), in the inferior aspect of the atrial septum; superior and inferior sinus venosus defects (in which the interatrial septum is intact), which are present at the site of the entry of the superior and inferior vena cava, respectively, into the right atrium; unroofed coronary sinus, the least common type.[Figure caption and citation for the preceding image starts]: Subtypes of atrial septal defects: (A) sinus venosus; (B) ostium secundum; (C) ostium primum; (D) unroofed coronary sinusMayo Clinic Foundation [Citation ends].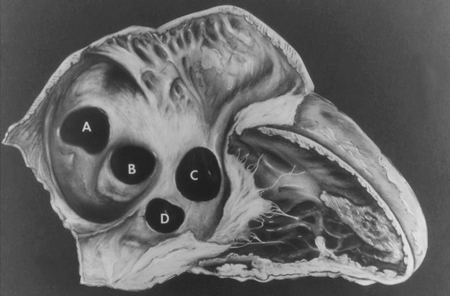 [Figure caption and citation for the preceding image starts]: Apical 4-chamber echocardiographic image of an ostium primum ASD (arrow). (RA) right atrium; (LA) left atrium; (LV) left ventricleImage courtesy of Patrick W. O'Leary, MD [Citation ends].
[Figure caption and citation for the preceding image starts]: Apical 4-chamber echocardiographic image of an ostium primum ASD (arrow). (RA) right atrium; (LA) left atrium; (LV) left ventricleImage courtesy of Patrick W. O'Leary, MD [Citation ends].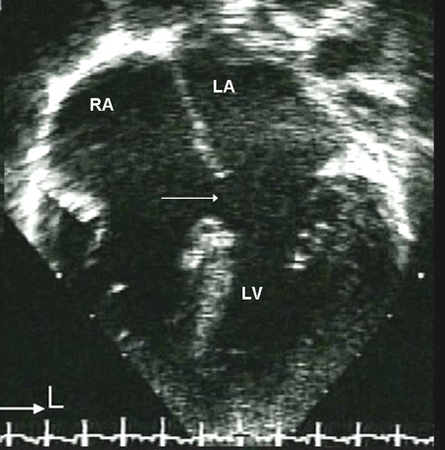
Unroofed coronary sinus is not a true atrial septal defect. It occurs when the roofing process of the coronary sinus in fetal life is incomplete, thus allowing the coronary sinus and left atrium to communicate.
Children with isolated pre-tricuspid shunts are frequently asymptomatic.[28] It is the most commonly missed CHD diagnosis in childhood, frequently not discovered until adulthood. Untreated defects of moderate to large size lead to exercise intolerance, atrial arrhythmias, and increased pulmonary blood flow. An actual increase in pulmonary vascular resistance is much more rare than in post-tricuspid shunts and occurs later in life, more commonly in specific sub-populations such as those with Down's syndrome. Paradoxical embolisation through the defect occurs almost exclusively in patients with an ASD secundum.
In patients with moderate to large defects, examination reveals an increased right ventricular impulse, a widely split fixed second heart sound, and a soft systolic ejection murmur best heard at the upper left sternal border, which is caused by increased blood flow through the pulmonary valve, rather than through the ASD. In large defects, a diastolic murmur may also be present at the lower sternal border secondary to excessive blood flow through the tricuspid valve.[28]
CXR is not helpful in determining the subtype and may be normal with a small ASD. ECG may also be normal in small secundum, sinus venous, and unroofed coronary sinus ASDs. With a larger shunt, there may be right atrial enlargement, right ventricular hypertrophy, or right axis deviation.[Figure caption and citation for the preceding image starts]: Parasternal short axis echocardiographic image demonstrating right ventricular enlargement in a patient with an ASD. (RV) right ventricle; (LV) left ventricleImage courtesy of Patrick W. O'Leary, MD [Citation ends].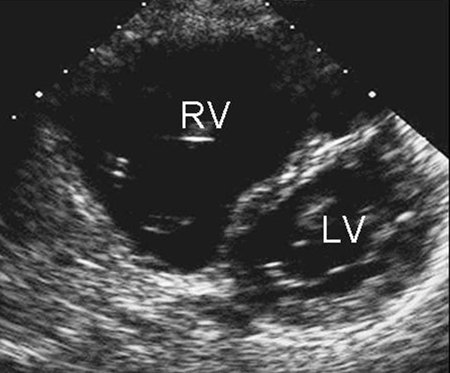 [Figure caption and citation for the preceding image starts]: Apical 4-chamber echocardiographic image demonstrating right ventricular enlargement in a patient with an ASD. (RA) right atrium; (RV) right ventricle; (LV) left ventricleImage courtesy of Patrick W. O'Leary, MD [Citation ends].
[Figure caption and citation for the preceding image starts]: Apical 4-chamber echocardiographic image demonstrating right ventricular enlargement in a patient with an ASD. (RA) right atrium; (RV) right ventricle; (LV) left ventricleImage courtesy of Patrick W. O'Leary, MD [Citation ends].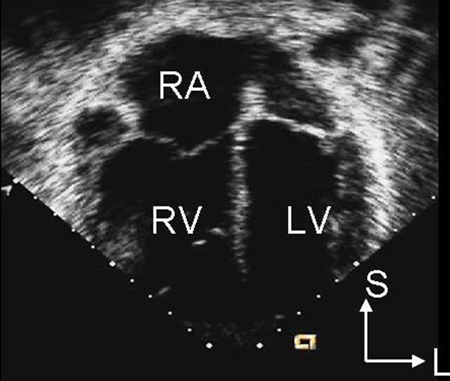 Ostium primum-type defects are characterised by an initial counter-clockwise frontal plane loop and left axis deviation.[28]
Ostium primum-type defects are characterised by an initial counter-clockwise frontal plane loop and left axis deviation.[28]
Treatment involves either an operation or percutaneous device closure.[29][Figure caption and citation for the preceding image starts]: Transesophageal echocardiographic image of an ASD occluder device (arrow). (RA) right atrium; (LA) left atrium; (SVC) superior vena cavaImage courtesy of Patrick W. O'Leary, MD [Citation ends].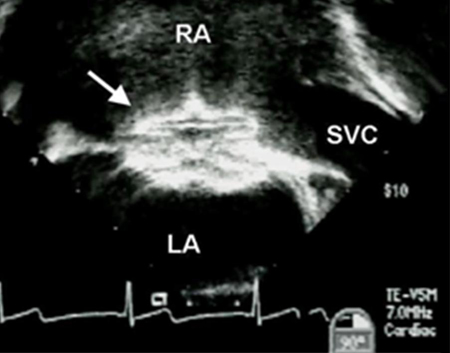 Device closure is the preferred method for ostium secundum defects if the defect is of adequate size (the largest closure device available is 40 mm in diameter) and if there are adequate septal rims to secure the device.[30] In most other subtypes, device closure is not feasible due to lack of rims and proximity to other cardiac structures, although recent reports suggest successful device closure of anatomically fit superior sinus venosus defects.[31] Partial AVSD (ostium primum ASD and a cleft mitral valve) can be repaired after the age of 18-24 months in most patients and involves closure of the mitral cleft in addition to the ASD.
Device closure is the preferred method for ostium secundum defects if the defect is of adequate size (the largest closure device available is 40 mm in diameter) and if there are adequate septal rims to secure the device.[30] In most other subtypes, device closure is not feasible due to lack of rims and proximity to other cardiac structures, although recent reports suggest successful device closure of anatomically fit superior sinus venosus defects.[31] Partial AVSD (ostium primum ASD and a cleft mitral valve) can be repaired after the age of 18-24 months in most patients and involves closure of the mitral cleft in addition to the ASD.
For more information see Interatrial communications.
Represents 4% to 5% of all CHDs and is found in 40% of children with Down's syndrome.[32] This defect is also called endocardial cushion defect or atrioventricular (AV) canal defect. Endocardial cushions close the ostium primum and form portions of the AV valves and the ventricular septum. AVSDs may be partial or complete; 1 type of a partial AVSD is a primum atrial septal defect (ASD). A complete AVSD consists of a primum ASD and a contiguous inlet ventricular septal defect (VSD).[Figure caption and citation for the preceding image starts]: Apical 4-chamber echocardiographic image of complete AVSD. Note the ostium primum ASD (*) and the contiguous inlet VSD (arrow). (RA) right atrium; (LA) left atrium; (RV) right ventricle; (LV) left ventricleImage courtesy of Patrick W. O'Leary, MD [Citation ends].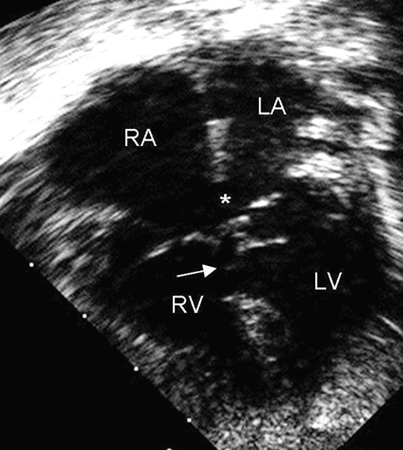
Presentation and findings on examination of patients with complete and partial AVSDs are similar to patients with a VSD or ASD, respectively. Severity is also determined by the volume of the shunt, extent of the AV valve abnormalities, associated cardiac and extra-cardiac anomalies, and the relative size of the 2 ventricles.
ECG is likely to reveal an initial counter-clockwise frontal plane loop and a superior QRS axis.[Figure caption and citation for the preceding image starts]: 12-lead ECG in an infant with complete AVSD; the ECG is significant for left axis deviationMayo Clinic Foundation [Citation ends].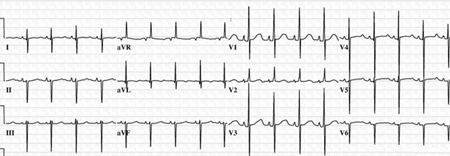 Patients with complete AVSDs usually have cardiomegaly and increased pulmonary vascular markings on CXR.[33]
Patients with complete AVSDs usually have cardiomegaly and increased pulmonary vascular markings on CXR.[33]
Children with a complete AVSD require surgical repair, usually performed at 3-6 months of age. They require life-long follow-up, as approximately 15% of patients develop progressive AV valve regurgitation or left ventricle outflow tract obstruction.[14]
A ductus arterious normally closes within several days of birth and is an essential component of normal in utero cardiovascular physiology. Persistent patency occurs in 1 in 2000 live births in full-term infants but is much more common in pre-term neonates.[34] In pre-term infants, a PDA has been noted during admission to hospital in 42% of neonates weighing 500-999 g, 21% weighing 1000-1499 g, and 7% weighing 1500-1750 g.[35] There is also a 30-fold higher incidence in patients born at higher altitudes. PDA represents 9% to 12% of all CHD.
Patients are asymptomatic when the ductus is small. With increasing size, newborns present with signs of increased pulmonary blood flow, a wide pulse pressure, and 'bounding' pulses. Eisenmenger's physiology, secondary to elevated pulmonary vascular resistance and shunt reversal, may occur if the PDA is large and long standing, and initially results in cyanosis only in the lower half of the body with gradual increase in systemic mixing and cyanosis as the disease progresses.[24]
Typically, a PDA produces a continuous murmur, best heard at the left infraclavicular area. The murmur is continuous because the aortic pressure is higher than the pulmonary artery pressure during both systole and diastole. If the shunt is large, a diastolic mitral flow murmur also may be heard. In some pre-term and newborn infants, the murmur may be heard only in systole and can be confused with the murmur of a VSD.
ECG and CXR often are normal. In a large PDA, bi-ventricular hypertrophy on the surface ECG, and increased pulmonary blood flow and cardiomegaly on CXR may be present. Echocardiography allows delineation of the PDA anatomy, and direction and volume of the shunt.[36] Further assessment of the anatomy and proximity to other structures can be obtained with cross-sectional laboratory such as a computed tomography study.[26]
In pre-term infants, the ductus can often be closed with indomethacin or ibuprofen, or failing that, ligated surgically.[37] In older children and adults, in the absence of significant pulmonary arterial hypertension, PDAs are closed percutaneously using coils or devices.[14][30] Diuretics can be used to treat patients with excess pulmonary blood flow until definitive closure.
For more information see Patent ductus arteriosus.
An anomaly where ≥1 (but not all) pulmonary veins connect to the systemic veins or right atrium, instead of to the left atrium. This defect represents <1% of all CHD. Anomalous connection of the right upper/middle pulmonary vein(s) is associated with a sinus venosus defect; connection of the right lower pulmonary vein(s) to the inferior vena cava may be part of an inferior sinus venosus defect, also referred to as 'Scimitar syndrome'.[38]
Presentation, ECG, and CXR findings are similar to patients with ASDs, although patients with only one abnormal vein are not infrequently clinically asymptomatic. The clinical presentation of patients with Scimitar syndrome varies from relatively asymptomatic adults to severely ill infants with ipsilateral lung hypoplasia, respiratory tract infections, and pulmonary hypertension.[39]
Trans-thoracic echocardiography may be insufficient to diagnose PAPVC accurately in adults, and other imaging modalities such as trans-oesophageal echocardiography, CT, or MRI may be necessary.
Surgical intervention is indicated if the shunt volume creates significant right-sided volume overload, which is associated with a pulmonary outflow murmur and occasionally a diastolic tricuspid inflow murmur.[28]
TOF is the most common cyanotic CHD, representing 4% to 8% of all defects, and can occur as a solitary cardiac lesion, or as part of a genetic syndrome, the most common of which is DiGeorge syndrome (a microdeletion of 22q11.2).[40] It consists of 4 abnormalities: outlet ventricular septal defect (VSD), right ventricular outflow tract (RVOT) obstruction, an aorta that over-rides the crest of the ventricular septum, and RV hypertrophy, a result of RVOT obstruction.[Figure caption and citation for the preceding image starts]: Parasternal long axis echocardiographic image in a patient with tetralogy of Fallot. The aorta (Ao) overrides the VSD (*). (LA) left atrium; (RV) right ventricle; (LV) left ventricleImage courtesy of Patrick W. O'Leary, MD [Citation ends].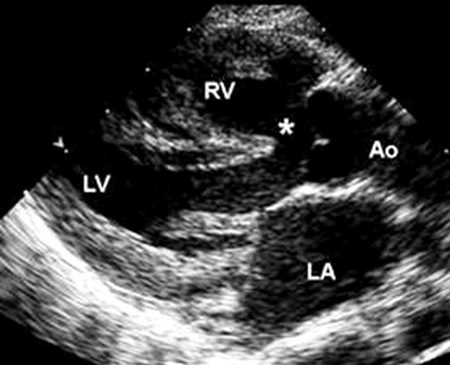
If RVOT obstruction is mild, mild or no right-to-left shunt occurs and there is no apparent cyanosis, a physiological setting sometimes referred to as 'pink tetralogy'. Most patients with TOF, however, have progressive cyanosis after birth followed by dyspnoea on exertion as a young child. Secondary erythrocytosis develops as a compensatory mechanism for chronic hypoxaemia. Poor somatic growth may be present. Hyper-cyanotic or 'tet spells' are hypoxic episodes consisting of an abrupt onset of rapid shallow breathing, increased agitation, cyanosis, and a decrease in murmur intensity due to reduced blood flow through the RVOT. Without treatment, these can cause profound hypoxaemia, seizures, and death. Older children with hypoxic spells will squat to increase systemic vascular resistance and thereby reduce hypoxaemia.
A loud ejection systolic murmur is heard at the left upper sternal border with radiation to both axillae due to the RVOT obstruction. Rarely, patients with minimal RVOT obstruction present with only mild cyanosis and may develop signs of excess pulmonary blood flow due to a left-to-right shunt through the VSD. A right ventricular impulse may be present.
Classically, CXR is normal except for decreased pulmonary vascular markings. Fewer than 25% of infants have the classically described 'boot-shaped' cardiac silhouette.
Severely hypoxic infants may be treated with prostaglandin E1 to re-open or maintain the patency of the ductus arteriosus and increase pulmonary blood flow. If this does not improve the hypoxaemia, the patient may require a systemic-to-pulmonary artery graft, such as a modified Blalock-Taussig shunt, until definitive repair (which includes patch closure of the VSD and relief of RVOT obstruction).[41] Complete repair may be performed in certain centres in the newborn period, although most clinicians favour it at 3 months of age or when progressive cyanosis develops. Surgical results have been excellent in recent decades, although many patients repaired as young children require re-implantation of a competent pulmonary valve when older, which can be performed either surgically or in a trans-catheter procedure.[14]
Follow-up laboratory is essential to identify timely need for valve replacement, type of treatment required, and to monitor treatment outcome.[42] Ventricular arrhythmias are a potential life risking long-term complication of repaired TOF, with the likely aetiology being slowly conducting anatomical isthmuses created at the time of the original surgical repair. Treatment includes ablation with or without intracardiac cardioverter defibrillator.[43]
For more information see Tetralogy of Fallot.
This often is considered to be the most severe form of tetralogy of Fallot where the pulmonary valve does not develop. Seventy per cent of patients have a patent ductus arteriosus (PDA) that maintains pulmonary blood flow. Others have multiple systemic-to-pulmonary collateral vessels. Pulmonary artery anomalies are common and include hypoplasia, non-confluence, and abnormal distribution.[44]
Patients are usually cyanotic at birth. A continuous murmur from the PDA or systemic-to-pulmonary artery collaterals may be audible. The second heart sound is single. ECG shows right axis deviation and right ventricular hypertrophy. CXR reveals markedly decreased pulmonary vascularity.
Prostaglandin E1 is commenced to maintain patency of the ductus arteriosus. Most patients are also likely to require a systemic-to-pulmonary shunt to maintain adequate pulmonary blood flow. Patients with large, confluent pulmonary arteries may have a single-stage repair; others require a staged repair. Patients with multiple systemic-to-pulmonary artery collaterals are at risk of developing early pulmonary hypertension, which initially can be segmental.[24]
This defect is rare and severity depends upon commonly associated right ventricular and tricuspid valve hypoplasia. Coronary artery-to-right ventricle fistulae are also associated with this defect.[45] A large, unrestrictive atrial septal defect (ASD) is required for survival as there is no outlet from the right ventricle to the pulmonary arteries.
There are no examination findings characteristic of this defect. CXR reveals cardiomegaly with a prominent right atrium and ECG shows atrial enlargement. Echocardiography is utilised to determine right ventricular size and tricuspid valve hypoplasia.
A restrictive ASD is a medical emergency in these patients and requires an urgent balloon atrial septostomy. Patients should be treated with prostaglandin E1 to maintain ductal patency because the patent ductus arteriosus is the only source of pulmonary blood flow.[46][47]
Most patients require staged operations that potentially allow the tricuspid valve and right ventricle to increase in size. A modified Fontan's operation or a 1.5 ventricle repair may be necessary if the right ventricle and tricuspid valve do not reach an adequate size to allow a 2 ventricle repair. A 1.5 ventricle repair involves a bi-directional cavopulmonary shunt ('Glenn' anastomosis) to divert superior vena caval blood flow to the pulmonary arteries to reduce right ventricular pre-load, closure of the ASD, and establishment of right ventricle-to-pulmonary artery continuity. Patients with coronary artery-to-right ventricle fistulae may require a modified Fontan's operation.
A common CHD representing 4% of all defects. In this defect, the aorta arises from the right ventricle and the pulmonary artery arises from the left ventricle. As a result, de-oxygenated systemic venous blood travels from the right ventricle to the aorta without passing through the lungs, and pulmonary venous blood travels from the left ventricle to the lungs via the pulmonary artery. The systemic and pulmonary circulations are in parallel rather than series. This is not compatible with life without a communication between the 2 circuits, such as a ventricular septal defect (VSD), atrial septal defect (ASD), or patent ductus arteriosus (PDA). Most patients with d-TGA do not have a VSD and a third of those with a VSD have associated pulmonary stenosis (PS).
Patients present in the first few hours of birth with cyanosis as a medical emergency.
Examination reveals a prominent right ventricular impulse, a single loud second heart sound (due to the anterior position of the aorta), and no murmur, or a systolic ejection murmur in those with a VSD and pulmonary stenosis.
Prostaglandin E1 is used to maintain patency of the ductus arteriosus and most patients also require an atrial septostomy to enhance mixing at the atrial level. The arterial switch operation, including coronary artery button re-implantation is the best option for patients with d-TGA and a normal pulmonary valve, and is performed in the first weeks of life. Once the pulmonary vascular resistance decreases, the left ventricle is no longer conditioned to pump against the higher resistance associated with the systemic circulation. TGA-VSD should be repaired in the newborn period if identified at that time. However, successful late repair (1-2 months of age) has been reported, albeit with increased morbidity. Patients with the combination of TGA, VSD, and PS may have undergone a Rastelli operation. This operation uses an angled VSD patch to direct blood from the left ventricle (LV) across the VSD to the anterior aorta. This procedure also involves placement of a conduit from the right ventricle (RV) to the pulmonary artery (PA). Although this operation results in restoration of the LV as the systemic ventricle, long-term management often involves multiple reoperations for replacement of the RV-to-PA conduit. Although no longer used, mainly older adults (>35-40 years of age) born with D-TGA have undergone the atrial switch procedure, either in the form of the Senning or the Mustard operation.[42] The RV is the systemic ventricle after these procedures.[30] Around 80% of these patients have had good outcome several decades after surgery. However, long-term complications, including arrhythmias and conduction abnormalities, systemic or pulmonary venous baffle obstruction or regurgitation, and systemic right ventricular dysfunction resulting in heart failure, occur in the vast majority of adult patients.[14] Adults presenting with RV dysfunction post atrial switch procedures are candidates for heart transplant.[30]
Comprises <1% of all CHD. It consists of a large outlet ventricular septal defect (VSD) and only 1 great artery, the truncus, arising from the heart, which gives rise to the coronary arteries, the pulmonary arteries, and the aorta. Four subtypes exist, based on the presence or absence of a main pulmonary artery, the branching of the pulmonary arteries and the formation of the aorta.[48]
[Figure caption and citation for the preceding image starts]: Subtypes of truncus arteriosus with ventricular septal defect (type A): Type A1: main pulmonary artery is present and bifurcates into the left and right pulmonary arteries Type A2:right and left branch pulmonary arteries arise from a common trunk. Type A3: One branch pulmonary artery arises from the common trunk and the other is absent/arises from a PDA or the aorta. Type A4: Truncus with aortic arch hypoplasia, coarctation or interrupted aortic arch and a large PDACalder L et al. Am Heart J 1976 Jul;92(1):23-38; used with permission [Citation ends].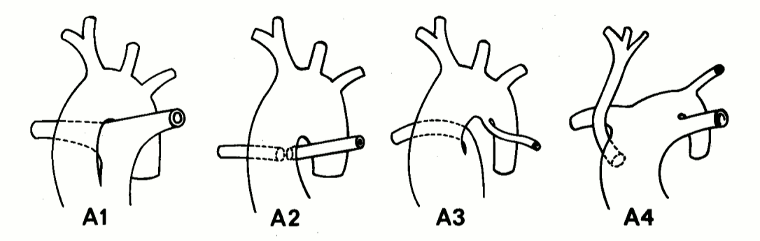
A micro-deletion of 22q11.2 (DiGeorge syndrome), interrupted aortic arch, and truncal valve insufficiency are common associated anomalies.
Patients present with congestive heart failure (CHF) or cyanosis.
Examination reveals a prominent right ventricle impulse and a systolic ejection murmur at the left sternal border. There may also be a truncal valve ejection click. Truncal valve regurgitation results in a decrescendo diastolic murmur. Interruption of the aorta and a restrictive PDA results in decreased femoral pulses.
Right ventricular dominance of infancy is seen on ECG, and CXR reveals cardiomegaly with increased pulmonary markings. Trans-thoracic echocardiography helps accurately identify various aspects of this lesion.[3][49][50]
CHF is treatable with diuretics. Surgical repair entails closure of the VSD, separation of the pulmonary and systemic circulations, and placement of a right ventricle-to-pulmonary artery conduit. Surgery is typically performed by 2-3 months of age to prevent the development of pulmonary hypertension with elevated pulmonary vascular resistance. Patients may also require multiple trans-catheter interventions or re-operations to treat dysfunction of the right ventricle-to-pulmonary conduit.[51] Operative mortality is greater with associated anomalies, such as interruption of the aortic arch.
A rare malformation of the tricuspid valve in which abnormal delamination of the septal and posterior valve leaflets leads to various degrees of tricuspid regurgitation, which can be severe at an early stage of life.[52][53][54] As a result of the lack of delamination and depending on its severity, some part of the right ventricle (RV) becomes 'atrialise', meaning that it lies below the level of the deformed tricuspid valve, and does not contribute to the forward flow. In rare cases, left ventricular (LV) myopathy is also present. In 80% to 94% of patients, a concomitant interatrial communication is present.[54] Cyanosis may occur due to streaming of deoxygenated blood from the eccentric tricuspid valve regurgitation jet and through the interatrial communication.
Patients have split first and second heart sounds. CXR reveals a markedly enlarged heart with a prominent right-sided border and decreased pulmonary vascularity. ECG shows tall 'p' waves in limb lead II and a right bundle branch block pattern.[54][55] Wolff Parkinson White syndrome is found in at least 15% of patients. Echocardiography, both trans-thoracic and trans-oesophageal assist in evaluating the valvular structural abnormalities and atrialised RV.[3] Cardiac MRI is helpful in delineating the degree of RV atrialisation, its size, and its function.
There is a wide range of anatomical and physiological severity. Patients with minimal cardiomegaly and tricuspid regurgitation lead normal lives without needing surgery. Newborns with severe forms of Ebstein's anomaly require prostaglandin E1 to augment pulmonary blood flow until pulmonary vascular resistance falls, and some do well without surgery. However, management may prove difficult in a subset of patients with massive cardiomegaly. Severe tricuspid regurgitation is an indication for surgery, after careful haemodynamic evaluation.[30]
Surgical interventions consist of tricuspid valve repair, replacement, or repair via Da Silva’s cone operation, which is a more extensive surgery in which the tricuspid valve is repaired, the annulus is returned to its anatomical location and excessive atrial tissue is resected. When performed by skilled operators, the results of the cone operation in terms of valve function are excellent.[56]
Comprising <1% of all CHD, TAPVC occurs when all the pulmonary veins connect to the systemic venous system or the right atrium. TAPVC can be sub-divided into supra-cardiac, cardiac, infra-cardiac, and mixed types, of which the supra-cardiac is the most common. TAPVC results in fully oxygenated blood from the lungs being directed back to the right atrium, and survival requires a defect in the atrial septum, without which blood cannot reach the aorta.[57] TAPVC can be complicated by obstruction of the veins that drain the pulmonary venous confluence, resulting in pulmonary hypertension and congestion.
Patients with unobstructed pulmonary veins and a large intra-atrial communication have mild cyanosis and increased pulmonary blood flow. Examination reveals a prominent right ventricle impulse and a systolic ejection murmur. A CXR shows moderate cardiomegaly and increased pulmonary vascular markings. The supra-cardiac form of TAPVC classically reveals a 'snowman' configuration. Echocardiography can show a dilated right atrium and right ventricle, and a posterior pulmonary venous confluence.[3]
Patients require surgery but have excellent long-term survival.
Patients with obstructed pulmonary venous blood flow present with respiratory distress secondary to pulmonary oedema. This is common in newborns who have an infra-cardiac connection, and patients require emergency surgical repair.[57] Mortality is as high as 40%, despite prompt surgery.
Results in the absence of a direct communication between the right atrium and right ventricle. This anomaly represents approximately 3% of all CHD. Initial survival is dependent on an intra-atrial communication. Presentation and treatment depend on the relationship of the great arteries (normal or transposed), presence of a ventricular septal defect (VSD), and severity of pulmonary valve stenosis.
Typically, patients present with cyanosis. The apical impulse may be prominent. The second heart sound may be single or split. A systolic murmur may be due to a VSD or pulmonary stenosis, while a mid-diastolic apical murmur may be due to increased pulmonary blood flow and congestive heart failure.
CXR reveals cardiomegaly with a prominent right atrial border. Pulmonary vascular markings vary depending on the degree of pulmonary stenosis. ECG is characterised by left axis deviation and an initial counter-clockwise frontal plane loop. There may be left ventricle hypertrophy.
If the source of pulmonary blood flow is not reliable, the patient should be treated with prostaglandin E1 until a systemic-to-pulmonary artery shunt can be established. Those with transposed great arteries have unrestricted pulmonary blood flow and require pulmonary artery banding to prevent pulmonary vascular obstructive disease and/or pulmonary oedema, along with anti-congestive medicines. All patients ultimately undergo a Fontan's operation in which the systemic venous return is connected directly to the pulmonary arteries (total cavo-pulmonary connection).[42] Long-term survival exceeds 85% at 10-year post-surgery follow-up. Long-term complications of the Fontan's operation include conduction abnormalities, arrhythmia, disorders of the lymphatics leading to plastic bronchitis and protein-losing enteropathy, congestive hepatopathy with liver fibrosis and ultimate cirrhosis and failure of the Fontan circulation warranting heart transplantation.
Refers to a spectrum of lesions that includes an intact ventricular septum with under-development of the left ventricle, and mitral and aortic valves. Patients commonly have coarctation of the aorta. Newborns can appear normal for several days after birth but are dependent on a patent ductus arteriosus (PDA). Once the ductus closes, they develop shock.
Examination reveals diminished lower extremity pulses and an increased right ventricular impulse. Echocardiography is used for pre- and post-operative assessment, although visualising the coronary artery anatomy can be a challenge due to the commonly associated aortic hypoplasia.[50]
Untreated, HLHS is lethal, although mortality has been markedly reduced in the last decade. Patients require a staged surgical approach.[58]
Stage 1 is a Norwood procedure, consisting of reconstruction of the aortic arch using the main pulmonary artery and a modified Blalock-Taussig shunt supplying blood flow to the pulmonary artery branches. Recently, placement of a right ventricle to pulmonary artery (Sano) shunt has been used as an alternative to a Blalock-Taussig shunt during stage 1 palliation.[58] Stage 2 consists of replacing the modified Blalock-Taussig shunt (or Sano shunt) with a bidirectional cavopulmonary (Glenn) anastomosis. Stage 3 consists of completion of the cavopulmonary connection and formation of the Fontan circulation, as detailed in the section on tricuspid atresia. In recent years, hybrid surgical/interventional catheterisation procedures have been utilised with branch pulmonary artery bands to restrict pulmonary blood flow, and stent deployment to maintain ductal patency, although the utility of this approach remains unclear. Cardiac transplantation is sometimes pursued as an alternative strategy to staged surgery.[59] Long-term complications of the Fontan circulation are similar to those described in the section on tricuspid atresia, and there is some evidence that in the presence of a systemic right ventricle these may occur at an earlier stage.
Includes 4 subtypes: aortic valve stenosis, supravalvular stenosis, discrete subvalvular stenosis, and tunnel subaortic stenosis.
Aortic valve stenosis is the most common and represents 5% of all CHD.[60] The leaflets or cusps are usually malformed or thickened. Approximately 2% of the general population has a bicuspid aortic valve, but many of these individuals never develop clinically significant aortic valve stenosis or regurgitation. Supravalvular stenosis occurs as an area of discrete or diffuse narrowing just distal to the sinotubular junction in the ascending aorta. It is frequently associated with Williams syndrome or a defect in the elastin gene on chromosome 7. Discrete subvalvular stenosis occurs when there is a fibromuscular ridge that causes obstruction just below the aortic valve. Turbulence of blood flow can cause damage to the valve leaflets of the aortic valve, resulting in progressive aortic regurgitation. Tunnel subaortic stenosis refers to a longer stenotic segment of the outflow tract when compared to discrete subvalvular stenosis.
Presentation depends on the severity of the obstruction. Most patients are asymptomatic. Older patients may present with chest pain or syncope. A subset of patients present with poor left ventricular function, low cardiac output, and signs of shock and congestive heart failure ('critical' aortic stenosis).
Examination reveals a crescendo-decrescendo systolic murmur, heard best at the left sternal border with radiation to the upper right sternal border. Moderate or severe stenosis results in a palpable thrill and delayed pulses. An ejection click may also be heard just after the first heart sound in patients with aortic valve stenosis. The click does not vary with respiration, in contrast to a pulmonary valve ejection click. Patients with concurrent aortic regurgitation also have a decrescendo diastolic murmur and a wide pulse pressure.
ECG inconsistently shows evidence of left ventricular hypertrophy. Severe LVOT stenosis is associated with flattening or inversion of the T waves in leads V5 and V6.
CXR may reveal a prominent aorta as a result of post-stenotic dilation. Echocardiography helps accurately evaluate aortic valve morphology and estimate the pressure gradient across the LVOT. In valvular aortic stenosis, the Doppler-estimated mean gradient is utilised to assess severity. The gradient will vary with the cardiac output.
Patients with 'critical' aortic stenosis require emergency relief of the stenosis. Symptomatic patients also require relief of the obstruction, regardless of the degree of the stenosis. Infants with severe valvular stenosis require balloon valvuloplasty, although surgery may be required for co-existent aortic regurgitation. Surgical valvotomy or valve replacement is considered for older children and adults. Patients with severe stenosis followed in the Second Natural History Study of Congenital Heart Defects had a 25-year survival rate of 81%.[61] Dilatation of the aorta, either at the level of the root or the ascending part, is present in 20% to 30% of patients with a bicuspid aortic valve, and in adult patients should be replaced when it reaches a diameter of 5.5 cm, or 5 cm if an aortic valve surgery is needed, or the patient has an inherited thoracic aortic aneurysm. [62]
Constitutes 5% of CHD and is more common in males; aortic coarction is associated with Turner's syndrome in females. It consists of a posterior ledge of tissue that protrudes into the aorta and is typically described as juxtaductal because it occurs across the ductus arteriosus. It is commonly associated with a bicuspid aortic valve (70% to 75% of patients with aortic coarctation have a bicuspid aortic valve, yet only about 7% of patients with bicuspid aortic valve also have aortic coarctation) and less commonly with a ventricular septal defect or subvalvular aortic stenosis.[63]
Patients with mild obstruction may not present until adolescence with a heart murmur or systemic hypertension. In those with severe obstruction, the patent ductus arteriosus (PDA) is the major source of systemic blood flow distal to the obstruction, which means that neonates develop metabolic acidosis and shock as the ductus closes.
The lower extremities have lower oxygen saturation and decreased pulses, in addition to congestive heart failure in patients with severe obstruction. It is important to take the blood pressure in both upper extremities and 1 leg before making the diagnosis as it is easy to miss the diagnosis in patients with a coarctation proximal to the left subclavian artery or those with an anomalous origin of the right subclavian artery. Turbulent blood flow through the area of coarctation produces a systolic murmur. Patients with an associated bicuspid aortic valve have an ejection click.
Infants, paradoxically, have right ventricular hypertrophy on ECG and echocardiography because of the PDA supplying the aorta distal to the obstruction. Older patients have signs of left ventricular hypertrophy. CT and MRI may be useful in defining the anatomy of the coarctation in stent implantation. The most commonly used surgical repair method is an extended end-to-end anastomosis, which when performed by skilled operators, has excellent duration and long-term outcomes.[64] Other surgical techniques include subclavian flap repair and use of a prosthetic graft. Repair in the newborn period is associated with an increased incidence of recurrence, which is best treated by stent placement. Other long-term complications include aneurysm formation at the site of the anastomosis and aortic dissection. Despite its localised nature, it is well appreciated that aortic coarctation causes systemic endothelial dysfunction, and many patients are at a risk of developing early-onset hypertension, which can lead to cardiovascular adverse events such as stroke, peripheral arterial disease, and ischaemic heart disease.[65] Up to 10% of patients develop intra-cranial vascular microaneurysms (sometimes referred to as 'berry aneurysms), especially in the anatomical area of the circle of Willis.[66] Life-long follow-up, even in patients who do not have a residual obstruction, is extremely important.
For more information see Aortic coarctation.
Represents up to 8% of all CHD and is commonly found in patients with Noonan syndrome. Most children present with an asymptomatic murmur. However, neonates with critical pulmonary valve stenosis present with cyanosis secondary to a right-to-left shunting of blood at the atrial level.[67]
Examination reveals an increased right ventricular impulse, a click following the first heart sound that varies with respiration, a normally split to a widely split second heart sound depending on the severity, and a crescendo-decrescendo ejection murmur. With increasing severity of stenosis, the pulmonary ejection click occurs earlier in systole; in the most severe cases, the click may merge with the first heart sound and become inaudible.[53] Conversely, the second heart sound splits more widely with increasing severity of stenosis and may become fixed in severe stenosis. In very severe stenosis, the pulmonary component of the second sound may become difficult to hear due to the loud murmur that spills into diastole. A fourth heart sound may be heard in patients with right ventricular failure.[53]
ECG may reveal right axis deviation and a CXR may show signs of right ventricular enlargement. Typically, there is post-stenotic dilation of the pulmonary arteries.
Balloon valvuloplasty is the preferred treatment, and at 10-year follow-up 84% of patients do not require further intervention.[67] The long-term outcome is excellent. If valve replacement is ultimately needed, it can be performed either surgically or via a trans-catheter approach.
For more information see Pulmonary stenosis.
People with CHD may be at increased risk for more severe COVID-19 infection, particularly those with more severe anatomical and physiological features of CHD.[68][69] Simple biventricular defects, complex ventricular defects, and heart surgery are associated with severe COVID-19.[70] Recommendations for prevention and management of COVID-19 in adults with CHD, based on risk stratification, have been published.[69][71]
It is recommended that cardiac medicines, including aspirin, angiotensin-converting enzyme (ACE) inhibitors, angiotensin-II receptor antagonists, beta-blockers, diuretics, and anti-arrhythmic medicines are continued during COVID-19 illness, unless there is a clear contraindication.[68]
For more information see Coronavirus disease 2019 (COVID-19).
Use of this content is subject to our disclaimer