History and exam
Key diagnostic factors
common
presence of risk factors
Many patients with X-linked or dominant retinitis pigmentosa (RP) will have FHx of RP. Autosomal recessive inheritance may or may not reveal FHx of retinal degeneration. RP is usually an isolated finding but can be a feature of some rare genetic disorders in combination with other symptoms such as balance problems, hearing loss, and renal disease.
positive family history
Inheritance can be autosomal dominant, autosomal recessive, X-linked, or, rarely, mitochondrial, or digenic. A family history can help to elicit the mode of inheritance. The prevalence of each type varies in different regions. Most patients who inherit genetic mutations will exhibit symptoms of the disease, but the severity can vary greatly, especially in autosomal dominant forms of the disease, which can show variable penetrance and expressivity.[27]
presence of an associated syndrome
Retinitis pigmentosa is usually an isolated finding but can be found as a feature of some rare genetic disorders in combination with other symptoms such as balance problems, hearing loss, and kidney disease. Syndromes include Usher's, Bardet-Biedl, Alstrom's, Joubert's, Senior-Loken, neuronal ceroid-lipofuscinosis, Kearns-Sayre, Bassen-Kornzweig disease (abetalipoproteinaemia), and infantile and adult Refsum's disease.
decreased peripheral vision
Key feature of the more common rod-cone form of retinitis pigmentosa. Visual field loss often goes unnoticed until it reaches a moderate stage due to the overlapping visual fields of each eye.
night blindness
Usually one of the earliest symptoms. Many patients report giving up driving at night due to difficulty seeing.
impaired dark adaptation
Patients often report difficulty entering movie theatres or, conversely, problems when going outside into a brightly lit environment.
decreased central acuity
Can be seen in advanced forms of the more common rod-cone dystrophy form or earlier if cystoid macular oedema develops. Cone-rod forms of retinitis pigmentosa can present with early decrease in central acuity.
atrophy of retinal pigment epithelium
A key feature. Depending on the mutation involved, can occur through primary retinal pigment epithelial degeneration or secondary to photoreceptor degeneration.
bone spicule pigmentation
A key feature, resulting from migration of retinal pigment epithelial cells into the photoreceptor layer of the retina, often surrounding retinal vessels. [Figure caption and citation for the preceding image starts]: Bone spiculesFrom the Oregon Retinal Degeneration Center collection [Citation ends].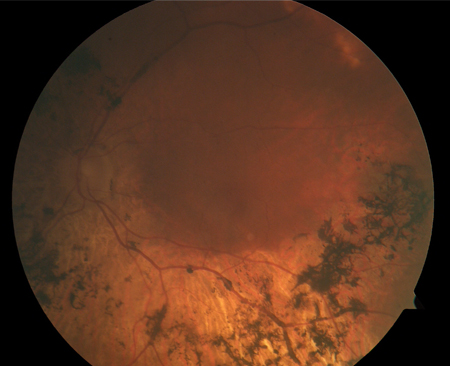
Other diagnostic factors
common
waxy pale optic nerve
Thought to be caused by gliosis around the optic nerve.[Figure caption and citation for the preceding image starts]: Waxy pallor and vascular attenuationFrom the Oregon Retinal Degeneration Center collection [Citation ends].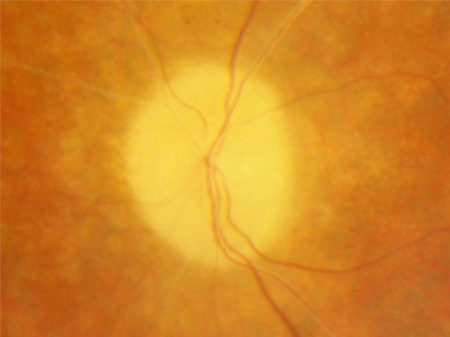
photopsias
Continual flashes of light, and are commonly experienced. It is important to distinguish these photopsias from flashes due to a retinal detachment or association with migraine.
refractive error
In early severe forms of retinitis pigmentosa, such as Leber's congenital amaurosis, hyperopia predominates. In later-onset forms, myopia and astigmatism are more common.
cataracts
Early development of posterior polar or subcapsular cataracts is especially common.
retinal vascular attenuation
Thought to be secondary to retinal atrophy. Its presence can help distinguish retinitis pigmentosa from choroideraemia, which is characterised by normal retinal vessels and atrophy of choroidal vessels. [Figure caption and citation for the preceding image starts]: Waxy pallor and vascular attenuationFrom the Oregon Retinal Degeneration Center collection [Citation ends].
cystoid macular oedema
A common complication. Optical coherence tomography is the best method of detection.
vitreous cells
Mild vitreous cells are often observed on slit-lamp examination of the fundus. More significant inflammation should prompt suspicion for other diseases that can mimic retinitis pigmentosa.
glare from bright lights
May be a problem in advanced disease.
uncommon
abnormal colour vision
Can be seen early in cone-rod dystrophies or later in rod-cone dystrophies. Usually affects the blue-yellow axis, which helps distinguish it from the more common X-linked red-green colour blindness, present in about 10% of the male population.
keratoconus
More common in patients with retinitis pigmentosa.
glaucoma
More common in patients with retinitis pigmentosa.[32]
optic nerve head drusen
Congenital and developmental anomalies of the optic nerve head. Formed by calcific degeneration in some optic nerve axons. Often found incidentally, but more common in patients with retinitis pigmentosa. With time can grow and impinge on the retinal nerve fibre layer and cause visual field defects. A small cup-to-disc ratio may indicate buried optic drusen.
Coats-like retinopathy
A rare sign in certain patients with retinitis pigmentosa. Characterised by peripheral areas of the retina with abnormal vascular telangiectasias, which can leak and cause retinal exudation.
Leber's congenital amaurosis
This is a severe subtype of retinitis pigmentosa, and can present in infancy with decreased vision, sluggish pupils, and nystagmus.[5]
Risk factors
strong
family history
Inheritance can be autosomal dominant, autosomal recessive, X-linked, or, rarely, mitochondrial or digenic. A family history can help to elicit the mode of inheritance. The prevalence of each type varies in different regions. Most patients who inherit genetic mutations will exhibit symptoms of the disease, but the severity can vary greatly, especially in autosomal dominant forms of the disease, which can show variable penetrance and expressivity.[27]
presence of an associated syndrome
Retinitis pigmentosa is usually an isolated finding but can be found as a feature of some rare genetic disorders in combination with other symptoms such as balance problems, hearing loss, and kidney disease. Syndromes include Usher's, Bardet-Biedl, Alstrom's, Joubert's, Senior-Loken, neuronal ceroid-lipofuscinosis, Kearns-Sayre, Bassen-Kornzweig disease (abetalipoproteinaemia), and infantile and adult Refsum's disease.[28][29]
Use of this content is subject to our disclaimer