The diagnostic approach includes obtaining a thorough history for key diagnostic symptoms such as a painless, unilateral upper abdominal/flank mass, congenital syndromes, and congenital urogenital anomalies.[4]Spreafico F, Fernandez CV, Brok J, et al. Wilms tumour. Nat Rev Dis Primers. 2021 Oct 14;7(1):75.
https://www.nature.com/articles/s41572-021-00308-8
http://www.ncbi.nlm.nih.gov/pubmed/34650095?tool=bestpractice.com
Abdominal ultrasound is the initial test of choice to establish the diagnosis of a renal tumour, and computed tomography (CT) or magnetic resonance imaging (MRI) of abdomen and pelvis are used to stage the tumour and plan further therapy.
Definitive diagnosis of suspected Wilms' tumour is based on histology of tumour following surgical resection (nephrectomy) or biopsy (if tumour is unresectable). Metastatic disease should be ruled out on CT chest (or chest x-ray in resource-limited areas) and abdominal/pelvis CT or MRI.
History
Family history of Wilms' tumour and presence of any congenital urogenital anomalies or a known predisposition syndrome should be documented.[4]Spreafico F, Fernandez CV, Brok J, et al. Wilms tumour. Nat Rev Dis Primers. 2021 Oct 14;7(1):75.
https://www.nature.com/articles/s41572-021-00308-8
http://www.ncbi.nlm.nih.gov/pubmed/34650095?tool=bestpractice.com
Any specific phenotypic anomalies that are associated with overgrowth or other genetic predisposition syndromes should be identified. For example, hyperinsulinaemic hypoglycaemia, which may be transient or persistent, occurs in 50% of children with Beckwith-Wiedemann syndrome during the neonatal period and infancy; therefore, if this syndrome is suspected, birth history for hypoglycaemia should be noted.[45]Green DM, Breslow NE, Beckwith JB, et al. Screening of children with hemihypertrophy, aniridia, and Beckwith-Wiedemann syndrome in patients with Wilms tumor: a report from the National Wilms Tumor Study. Med Pediatr Oncol. 1993;21(3):188-92.
http://www.ncbi.nlm.nih.gov/pubmed/8095320?tool=bestpractice.com
Epidemiological features may help; Wilms' tumour is more common in black and white children compared with Asian children, and most commonly occurs in the first 5 years of life.[1]Nakata K, Colombet M, Stiller CA, et al. Incidence of childhood renal tumours: an international population-based study. Int J Cancer. 2020 Dec 15;147(12):3313-27.
https://onlinelibrary.wiley.com/doi/10.1002/ijc.33147
http://www.ncbi.nlm.nih.gov/pubmed/32902866?tool=bestpractice.com
[16]National Cancer Institute. Statistics for cancers in children, adolescents, and young adults. Oct 2024 [internet publication].
https://nccrexplorer.ccdi.cancer.gov
Children usually present with a painless, unilateral upper abdominal/flank mass.[4]Spreafico F, Fernandez CV, Brok J, et al. Wilms tumour. Nat Rev Dis Primers. 2021 Oct 14;7(1):75.
https://www.nature.com/articles/s41572-021-00308-8
http://www.ncbi.nlm.nih.gov/pubmed/34650095?tool=bestpractice.com
Additional clinical features may include pallor, abdominal pain, fever, haematuria (visible or non-visible), poor appetite, and weight loss.[4]Spreafico F, Fernandez CV, Brok J, et al. Wilms tumour. Nat Rev Dis Primers. 2021 Oct 14;7(1):75.
https://www.nature.com/articles/s41572-021-00308-8
http://www.ncbi.nlm.nih.gov/pubmed/34650095?tool=bestpractice.com
Shortness of breath may be associated with anaemia, lung metastasis, or abdominal compression on the diaphragm.[4]Spreafico F, Fernandez CV, Brok J, et al. Wilms tumour. Nat Rev Dis Primers. 2021 Oct 14;7(1):75.
https://www.nature.com/articles/s41572-021-00308-8
http://www.ncbi.nlm.nih.gov/pubmed/34650095?tool=bestpractice.com
Physical examination
The location and extent of the abdominal mass should be documented, if possible. The mass is usually retroperitoneal ('ballotable'), and does not move with respirations.[4]Spreafico F, Fernandez CV, Brok J, et al. Wilms tumour. Nat Rev Dis Primers. 2021 Oct 14;7(1):75.
https://www.nature.com/articles/s41572-021-00308-8
http://www.ncbi.nlm.nih.gov/pubmed/34650095?tool=bestpractice.com
It is smooth, firm to touch, and non-tender. The child may have abdominal distention. Genitalia should be examined for any congenital urogenital anomalies (i.e., hypospadias, atypical genitalia, or cryptorchidism).[20]Narod SA, Hawkins MM, Robertson CM, et al. Congenital anomalies and childhood cancer in Great Britain. Am J Hum Genet. 1997 Mar;60(3):474-85.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1712528
http://www.ncbi.nlm.nih.gov/pubmed/9042906?tool=bestpractice.com
[21]Breslow NE, Beckwith JB. Epidemiological features of Wilms' tumor: results of the National Wilms' Tumor Study. J Natl Cancer Inst. 1982 Mar;68(3):429-36.
http://www.ncbi.nlm.nih.gov/pubmed/6278194?tool=bestpractice.com
Presence of a varicocele in supine position may be associated with tumour extension into the inferior vena cava or the renal vein.[46]Idowu BM, Tanimola AG. Wilm's tumor presenting with scrotal varicocele in an 11-month-old boy. Indian J Radiol Imaging. 2018 Apr-Jun;28(2):247-9.
https://www.thieme-connect.com/products/ejournals/abstract/10.4103/ijri.IJRI_279_17
http://www.ncbi.nlm.nih.gov/pubmed/30050251?tool=bestpractice.com
Hypertension is present in approximately 25% of patients, and is secondary to compression of renal vasculature, or due to renin hypersecretion.[5]Davidoff AM. Wilms tumor. Adv Pediatr. 2012;59(1):247-67.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3589819
http://www.ncbi.nlm.nih.gov/pubmed/22789581?tool=bestpractice.com
[47]Maas MH, Cransberg K, van Grotel M, et al. Renin-induced hypertension in Wilms tumor patients. Pediatr Blood Cancer. 2007 May;48(5):500-3.
http://www.ncbi.nlm.nih.gov/pubmed/16794999?tool=bestpractice.com
Hepatomegaly may indicate metastatic disease.[4]Spreafico F, Fernandez CV, Brok J, et al. Wilms tumour. Nat Rev Dis Primers. 2021 Oct 14;7(1):75.
https://www.nature.com/articles/s41572-021-00308-8
http://www.ncbi.nlm.nih.gov/pubmed/34650095?tool=bestpractice.com
Intracardiac extension of Wilms' tumour is less common.[11]Abdullah Y, Karpelowsky J, Davidson A, et al. Management of nine cases of Wilms' tumour with intracardiac extension - a single centre experience. J Pediatr Surg. 2013 Feb;48(2):394-9.
http://www.ncbi.nlm.nih.gov/pubmed/23414872?tool=bestpractice.com
Phenotypic abnormalities that may be characteristic of Wilms' tumour-predisposition syndromes should be documented.[4]Spreafico F, Fernandez CV, Brok J, et al. Wilms tumour. Nat Rev Dis Primers. 2021 Oct 14;7(1):75.
https://www.nature.com/articles/s41572-021-00308-8
http://www.ncbi.nlm.nih.gov/pubmed/34650095?tool=bestpractice.com
Very rarely, children may present with a paraneoplastic syndrome that affects the central and peripheral nervous system (e.g., generalised weakness, fatigue, ptosis, hypokinesis, dysarthria, urinary retention, facial diplegia, ophthalmoplegia, and autonomic dysfunction).[10]Petersen CL, Hemker BG, Jacobson RD, et al. Wilms tumor presenting with lambert-eaton myasthenic syndrome. J Pediatr Hematol Oncol. 2013 May;35(4):267-70.
http://www.ncbi.nlm.nih.gov/pubmed/23612377?tool=bestpractice.com
Laboratory investigations
Full blood count, renal and hepatic function, urinalysis, and coagulation studies are typically ordered, although they are not required for diagnosis.[48]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: Wilms tumor (nephroblastoma) [internet publication].
https://www.nccn.org/guidelines/category_1
Imaging
Initial studies are aimed at establishing renal origin and extent of the mass. Abdominal ultrasonography is the recommended first-line test for establishing presumptive diagnosis of a renal tumour.[4]Spreafico F, Fernandez CV, Brok J, et al. Wilms tumour. Nat Rev Dis Primers. 2021 Oct 14;7(1):75.
https://www.nature.com/articles/s41572-021-00308-8
http://www.ncbi.nlm.nih.gov/pubmed/34650095?tool=bestpractice.com
[49]van der Beek JN, Artunduaga M, Schenk JP, et al. Similarities and controversies in imaging of pediatric renal tumors: a SIOP-RTSG and COG collaboration. Pediatr Blood Cancer. 2022 Nov 9;e30080.
http://www.ncbi.nlm.nih.gov/pubmed/36349564?tool=bestpractice.com
Relatively easy to perform without sedation in young children, abdominal ultrasound establishes the renal origin, and the number and location of renal masses.[49]van der Beek JN, Artunduaga M, Schenk JP, et al. Similarities and controversies in imaging of pediatric renal tumors: a SIOP-RTSG and COG collaboration. Pediatr Blood Cancer. 2022 Nov 9;e30080.
http://www.ncbi.nlm.nih.gov/pubmed/36349564?tool=bestpractice.com
Typical findings are a large echogenic, heterogenous, unilateral, mainly solid (although small areas of cystic changes may be seen) intrarenal mass.
If Wilms' tumour is suspected on ultrasound, the patient should be promptly referred to a paediatric cancer centre for further evaluation and management.
Tumour staging
Either CT or MRI of the abdomen and pelvis should be obtained for locoregional staging by evaluating the contralateral kidney for synchronous disease and determining the size and number of ipsilateral masses, presence of lymphadenopathy, presence and extent of tumour thrombus, and presence of metastatic disease to organs such as the liver.[49]van der Beek JN, Artunduaga M, Schenk JP, et al. Similarities and controversies in imaging of pediatric renal tumors: a SIOP-RTSG and COG collaboration. Pediatr Blood Cancer. 2022 Nov 9;e30080.
http://www.ncbi.nlm.nih.gov/pubmed/36349564?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: Wilms' tumour: MRI findingsUHRAD.com; used with permission [Citation ends].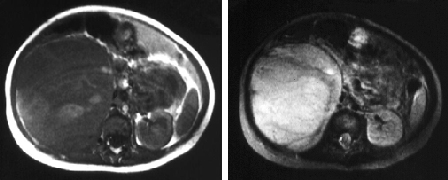
Signs of possible rupture and infiltration into adjacent organs may be observed; however, imaging has a poor predictive value for preoperative rupture.[50]Khanna G, Naranjo A, Hoffer F, et al. Detection of preoperative wilms tumor rupture with CT: a report from the Children's Oncology Group. Radiology. 2013 Feb;266(2):610-7.
https://pubs.rsna.org/doi/10.1148/radiol.12120670?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200pubmed
http://www.ncbi.nlm.nih.gov/pubmed/23192775?tool=bestpractice.com
The International Society of Paediatric Oncology (SIOP) protocol, and the Children's Oncology Group (COG) protocol, advocate chest CT for detection of lung lesions at diagnosis.[49]van der Beek JN, Artunduaga M, Schenk JP, et al. Similarities and controversies in imaging of pediatric renal tumors: a SIOP-RTSG and COG collaboration. Pediatr Blood Cancer. 2022 Nov 9;e30080.
http://www.ncbi.nlm.nih.gov/pubmed/36349564?tool=bestpractice.com
In resource-limited regions, chest x-ray can be used to identify lung metastasis; however, plain radiography may miss smaller pulmonary lesions (typically <1 cm).[4]Spreafico F, Fernandez CV, Brok J, et al. Wilms tumour. Nat Rev Dis Primers. 2021 Oct 14;7(1):75.
https://www.nature.com/articles/s41572-021-00308-8
http://www.ncbi.nlm.nih.gov/pubmed/34650095?tool=bestpractice.com
Ultrasound to assess venous involvement and additional urogenital tract imaging to assess for ureteric involvement is controversial and not routinely needed. The role of positron emission tomography scan in the staging and assessment of response is not established.[51]Provenzi M, Saettini F, Conter V, et al. Is there a role for FDG-PET for the assessment of treatment efficacy in Wilms' tumor? A case report and literature review. Pediatr Hematol Oncol. 2013 Oct;30(7):633-9.
http://www.ncbi.nlm.nih.gov/pubmed/24050763?tool=bestpractice.com
[Figure caption and citation for the preceding image starts]: Wilms' tumour: MRI findingsUHRAD.com; used with permission [Citation ends].
Tumour histology
Definitive diagnosis is based on histology of tumour following surgical resection (nephrectomy).[4]Spreafico F, Fernandez CV, Brok J, et al. Wilms tumour. Nat Rev Dis Primers. 2021 Oct 14;7(1):75.
https://www.nature.com/articles/s41572-021-00308-8
http://www.ncbi.nlm.nih.gov/pubmed/34650095?tool=bestpractice.com
[48]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: Wilms tumor (nephroblastoma) [internet publication].
https://www.nccn.org/guidelines/category_1
If the tumour is unresectable, the COG protocol recommends an open biopsy or core needle biopsy with a minimum of 10-12 non-necrotic cores to ensure sufficient tissue for molecular testing.[4]Spreafico F, Fernandez CV, Brok J, et al. Wilms tumour. Nat Rev Dis Primers. 2021 Oct 14;7(1):75.
https://www.nature.com/articles/s41572-021-00308-8
http://www.ncbi.nlm.nih.gov/pubmed/34650095?tool=bestpractice.com
[52]Lopyan NM, Ehrlich PF. Surgical management of Wilms tumor (nephroblastoma) and renal cell carcinoma in children and young adults. Surg Oncol Clin N Am. 2021 Apr;30(2):305-23.
http://www.ncbi.nlm.nih.gov/pubmed/33706902?tool=bestpractice.com
The SIOP protocol does not routinely recommend pretreatment biopsy.[53]Vujanić GM, Gessler M, Ooms AHAG, et al. The UMBRELLA SIOP-RTSG 2016 Wilms tumour pathology and molecular biology protocol. Nat Rev Urol. 2018 Nov;15(11):693-701.
https://www.nature.com/articles/s41585-018-0100-3
http://www.ncbi.nlm.nih.gov/pubmed/30310143?tool=bestpractice.com
Percutaneous cutting needle biopsy (tru-cut biopsy) can potentially be considered in patients whose tumours are suspected not to be Wilms' tumour, such as young children with stage IV disease and in children >10 years of age (as the frequency of non-Wilms renal tumours is increased in these patient populations).[53]Vujanić GM, Gessler M, Ooms AHAG, et al. The UMBRELLA SIOP-RTSG 2016 Wilms tumour pathology and molecular biology protocol. Nat Rev Urol. 2018 Nov;15(11):693-701.
https://www.nature.com/articles/s41585-018-0100-3
http://www.ncbi.nlm.nih.gov/pubmed/30310143?tool=bestpractice.com
[54]van den Heuvel-Eibrink MM, van Tinteren H, Rehorst H, et al. Malignant rhabdoid tumours of the kidney (MRTKs), registered on recent SIOP protocols from 1993 to 2005: a report of the SIOP renal tumour study group. Pediatr Blood Cancer. 2011 May;56(5):733-7.
http://www.ncbi.nlm.nih.gov/pubmed/21370404?tool=bestpractice.com
Genetic and molecular biomarker testing
Molecular genetic testing is a useful diagnostic tool for the identification of phenotypic syndromes that may be associated with Wilms' tumour.
Genetic testing for changes in WT1 and other genes associated with Wilms' tumour may be considered for all children with Wilms' tumour. In particular, genetic testing is recommended for patients at high risk for an underlyingpredisposition syndrome. Features suggestive of a predisposition syndrome include bilateral or multifocal disease, early onset (aged <2 years), family history of Wilms' tumour, multiple nephrogenic rests, unexplained proteinuria or renal failure, or urogenital or other phenotypic anomalies associated with predisposing syndromes.[48]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: Wilms tumor (nephroblastoma) [internet publication].
https://www.nccn.org/guidelines/category_1
[55]National Health Service. National Genomic Test Directory. Testing criteria for rare and inherited disease. May 2025 [internet publication].
https://www.england.nhs.uk/wp-content/uploads/2018/08/rare-inherited-disease-eligibility-criteria-v8.0.pdf
Gene panels are available for Wilms' tumour predisposition that include WT1, REST, TRIM28, and other relevant genes.[38]Brzezinski JJ, Becktell KD, Bougeard G, et al. Update on surveillance guidelines in emerging Wilms tumor predisposition syndromes. Clin Cancer Res. 2025 Jan 6;31(1):18-24.
https://aacrjournals.org/clincancerres/article/31/1/18/750711/Update-on-Surveillance-Guidelines-in-Emerging
http://www.ncbi.nlm.nih.gov/pubmed/39466169?tool=bestpractice.com
[48]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: Wilms tumor (nephroblastoma) [internet publication].
https://www.nccn.org/guidelines/category_1
[56]Genomics England PanelApp. Wilms tumour with features suggestive of predisposition (Version 1.3). Nov 2022 [internet publication].. Wilms tumour with features suggestive of predisposition (Version 1.3). Nov 2022 [internet publication].
https://panelapp.genomicsengland.co.uk/panels/1108
[57]Hol JA, Kuiper RP, van Dijk F, et al. Prevalence of (epi)genetic predisposing factors in a 5-year unselected national Wilms tumor cohort: a comprehensive clinical and genomic characterization. J Clin Oncol. 2022 Jun 10;40(17):1892-902.
https://ascopubs.org/doi/10.1200/JCO.21.02510?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200pubmed
http://www.ncbi.nlm.nih.gov/pubmed/35230882?tool=bestpractice.com
Patients with clinical features suggestive of Beckwith-Wiedemann syndrome may be tested for genetic and/or epigenetic changes at the 11p15.5 imprinted region, which may also be mosaic.[37]Kalish JM, Becktell KD, Bougeard G, et al. Update on surveillance for Wilms tumor and hepatoblastoma in Beckwith-Wiedemann syndrome and other predisposition Ssyndrome. Clin Cancer Res. 2024 Dec 2;30(23):5260-9.
https://aacrjournals.org/clincancerres/article/30/23/5260/750189/Update-on-Surveillance-for-Wilms-Tumor-and
http://www.ncbi.nlm.nih.gov/pubmed/39320341?tool=bestpractice.com
[38]Brzezinski JJ, Becktell KD, Bougeard G, et al. Update on surveillance guidelines in emerging Wilms tumor predisposition syndromes. Clin Cancer Res. 2025 Jan 6;31(1):18-24.
https://aacrjournals.org/clincancerres/article/31/1/18/750711/Update-on-Surveillance-Guidelines-in-Emerging
http://www.ncbi.nlm.nih.gov/pubmed/39466169?tool=bestpractice.com
[57]Hol JA, Kuiper RP, van Dijk F, et al. Prevalence of (epi)genetic predisposing factors in a 5-year unselected national Wilms tumor cohort: a comprehensive clinical and genomic characterization. J Clin Oncol. 2022 Jun 10;40(17):1892-902.
https://ascopubs.org/doi/10.1200/JCO.21.02510?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200pubmed
http://www.ncbi.nlm.nih.gov/pubmed/35230882?tool=bestpractice.com
Testing strategies may combine a Wilms' tumour gene panel with testing for Beckwith-Wiedemann syndrome, or may be targeted for patients with clinical features suggestive of a specific syndrome.[57]Hol JA, Kuiper RP, van Dijk F, et al. Prevalence of (epi)genetic predisposing factors in a 5-year unselected national Wilms tumor cohort: a comprehensive clinical and genomic characterization. J Clin Oncol. 2022 Jun 10;40(17):1892-902.
https://ascopubs.org/doi/10.1200/JCO.21.02510?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200pubmed
http://www.ncbi.nlm.nih.gov/pubmed/35230882?tool=bestpractice.com
Tumour molecular markers
Evaluated to help guide risk assessment and treatment for favourable risk Wilms' tumours.[48]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: Wilms tumor (nephroblastoma) [internet publication].
https://www.nccn.org/guidelines/category_1
Assays for biomarkers associated with unfavourable outcomes, such as loss of heterozygosity (LOH) in 16q, 11p, and 1p, and gain of chromosome 1q, are performed on tumour tissue.[33]Grundy PE, Breslow NE, Li S, et al. Loss of heterozygosity for chromosomes 1p and 16q is an adverse prognostic factor in favorable-histology Wilms tumor: a report from the National Wilms Tumor Study Group. J Clin Oncol. 2005 Oct 10;23(29):7312-21.
http://www.ncbi.nlm.nih.gov/pubmed/16129848?tool=bestpractice.com
[48]National Comprehensive Cancer Network. NCCN clinical practice guidelines in oncology: Wilms tumor (nephroblastoma) [internet publication].
https://www.nccn.org/guidelines/category_1
[58]Wittmann S, Zirn B, Alkassar M, et al. Loss of 11q and 16q in Wilms tumors is associated with anaplasia, tumor recurrence, and poor prognosis. Genes Chromosomes Cancer. 2007 Feb;46(2):163-70.
http://www.ncbi.nlm.nih.gov/pubmed/17099873?tool=bestpractice.com
[59]Gratias EJ, Dome JS, Jennings LJ, et al. Association of chromosome 1q gain with inferior survival in favorable-histology Wilms tumor: a report from the Children's Oncology Group. J Clin Oncol. 2016 Sep 10;34(26):3189-94.
https://ascopubs.org/doi/10.1200/JCO.2015.66.1140?url_ver=Z39.88-2003&rfr_id=ori:rid:crossref.org&rfr_dat=cr_pub%20%200pubmed
http://www.ncbi.nlm.nih.gov/pubmed/27400937?tool=bestpractice.com
Tumour biobanking
In every child with Wilms' tumour, biobanking of tumour material (fresh frozen tumour tissue) and healthy kidney tissue should be offered to facilitate research studies to define new risk factors and treatment targets.