A insuficiência adrenal primária (IAP) pode ser difícil de diagnosticar. As manifestações clínicas baseiam-se na deficiência de hormônios adrenocorticais (glicocorticoides, mineralcorticoides) e androgênios adrenais.[9]Betterle C, Presotto F, Furmaniak J. Epidemiology, pathogenesis, and diagnosis of Addison's disease in adults. J Endocrinol Invest. 2019 Dec;42(12):1407-33.
http://www.ncbi.nlm.nih.gov/pubmed/31321757?tool=bestpractice.com
Ela se manifesta com sinais e sintomas inespecíficos, como fadiga, fraqueza e perda de peso, que são comuns a outras doenças. Outras manifestações incluem hiperpigmentação, hipotensão postural devido à perda de sal e à desidratação, hiponatremia, hipercalemia, alterações no hemograma (anemia, eosinofilia e linfocitose) e hipoglicemia.[2]Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2016 Feb;101(2):364-89.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4880116
http://www.ncbi.nlm.nih.gov/pubmed/26760044?tool=bestpractice.com
Uma alta porcentagem de pacientes é diagnosticada após uma crise adrenal com risco de vida.[2]Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2016 Feb;101(2):364-89.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4880116
http://www.ncbi.nlm.nih.gov/pubmed/26760044?tool=bestpractice.com
Os pacientes que apresentam características de crise adrenal (ou seja, hipotensão, insuficiência circulatória) devem ser tratados com urgência.[26]Arlt W; Society for Endocrinology Clinical Committee. Society For Endocrinology endocrine emergency guidance: emergency management of acute adrenal insufficiency (adrenal crisis) in adult patients. Endocr Connect. 2016 Sep;5(5):G1-3.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5314805
http://www.ncbi.nlm.nih.gov/pubmed/27935813?tool=bestpractice.com
Devem ser realizados exames como linha basal, mas os exames diagnósticos não devem protelar o tratamento.[26]Arlt W; Society for Endocrinology Clinical Committee. Society For Endocrinology endocrine emergency guidance: emergency management of acute adrenal insufficiency (adrenal crisis) in adult patients. Endocr Connect. 2016 Sep;5(5):G1-3.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5314805
http://www.ncbi.nlm.nih.gov/pubmed/27935813?tool=bestpractice.com
Os grupos de pacientes com fatores causativos de desenvolvimento de IAP devem ser considerados de alto risco, de modo que um diagnóstico possa ser estabelecido precocemente. Os fatores de risco incluem sexo feminino e doenças autoimunes conhecidas, como diabetes do tipo 1, artrite reumatoide, anemia perniciosa, doença celíaca, vitiligo ou doença tireoidiana autoimune.[3]Husebye ES, Pearce SH, Krone NP, et al. Adrenal insufficiency. Lancet. 2021 Feb 13;397(10274):613-29.
http://www.ncbi.nlm.nih.gov/pubmed/33484633?tool=bestpractice.com
[9]Betterle C, Presotto F, Furmaniak J. Epidemiology, pathogenesis, and diagnosis of Addison's disease in adults. J Endocrinol Invest. 2019 Dec;42(12):1407-33.
http://www.ncbi.nlm.nih.gov/pubmed/31321757?tool=bestpractice.com
[13]Dalin F, Nordling Eriksson G, Dahlqvist P, et al. Clinical and immunological characteristics of autoimmune Addison disease: a nationwide Swedish multicenter study. J Clin Endocrinol Metab. 2017 Feb 1;102(2):379-89.
https://www.doi.org/10.1210/jc.2016-2522
http://www.ncbi.nlm.nih.gov/pubmed/27870550?tool=bestpractice.com
Outro fator de risco para o desenvolvimento de IAP é a ocorrência de hemorragia adrenal ou infarto hemorrágico. Isso pode ser causado por estados tromboembólicos e/ou hipercoaguláveis, como síndrome antifosfolipídica, sepse e trombocitopenia induzida por heparina.[9]Betterle C, Presotto F, Furmaniak J. Epidemiology, pathogenesis, and diagnosis of Addison's disease in adults. J Endocrinol Invest. 2019 Dec;42(12):1407-33.
http://www.ncbi.nlm.nih.gov/pubmed/31321757?tool=bestpractice.com
[17]Rosenberger LH, Smith PW, Sawyer RG, et al. Bilateral adrenal hemorrhage: the unrecognized cause of hemodynamic collapse associated with heparin-induced thrombocytopenia. Crit Care Med. 2011 Apr;39(4):833-8.
http://www.ncbi.nlm.nih.gov/pubmed/21242799?tool=bestpractice.com
Pacientes que tomam anticoagulantes apresentam aumento do risco de hemorragia adrenal.[2]Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2016 Feb;101(2):364-89.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4880116
http://www.ncbi.nlm.nih.gov/pubmed/26760044?tool=bestpractice.com
A insuficiência adrenal secundária pode estar associada à deficiência grave de cortisol, mas tem um quadro clínico diferente (por exemplo, sem aumento da pigmentação e somente com discreta deficiência de mineralocorticoide). Portanto, o achado de hipocortisolismo não é exclusivo da IAP. Uma história de tratamento com glicocorticoide nos últimos meses, ou a possibilidade de doença hipotalâmica-hipofisária (por exemplo, surgimento simultâneo de outras deficiências hipofisárias secundárias, como as deficiências da tireoide ou gonadais), deve justificar a avaliação para insuficiência adrenal secundária.
História
Fadiga e letargia (ou fraqueza) são os sintomas mais comumente presentes.[3]Husebye ES, Pearce SH, Krone NP, et al. Adrenal insufficiency. Lancet. 2021 Feb 13;397(10274):613-29.
http://www.ncbi.nlm.nih.gov/pubmed/33484633?tool=bestpractice.com
Eles são geralmente insidiosos e podem estar acompanhados por vários graus de fraqueza muscular generalizada, mialgias e artralgias. Os pacientes reclamam de perda gradual da energia ao longo do dia.
Anorexia está frequentemente presente e pode levar à perda de peso significativa. Ambos os sintomas são características importantes em pacientes com IAP.
Tontura, artralgia e mialgia são menos comuns.
Sintomas gastrointestinais, inclusive náuseas, vômitos e dor abdominal vaga são sintoma de apresentação inespecíficos e podem sugerir crise adrenal se houver agravamento agudo.[3]Husebye ES, Pearce SH, Krone NP, et al. Adrenal insufficiency. Lancet. 2021 Feb 13;397(10274):613-29.
http://www.ncbi.nlm.nih.gov/pubmed/33484633?tool=bestpractice.com
A fissura por sal pode estar presente.[3]Husebye ES, Pearce SH, Krone NP, et al. Adrenal insufficiency. Lancet. 2021 Feb 13;397(10274):613-29.
http://www.ncbi.nlm.nih.gov/pubmed/33484633?tool=bestpractice.com
A história deve ser direcionada para estabelecer a causa subjacente, como uma história conhecida de doença autoimune, infecção pelo vírus da imunodeficiência humana (HIV), possível infecção por tuberculose e medicamentos em uso na atualidade.
Alterações súbitas no controle glicêmico e hipoglicemia recorrente em pacientes com diabetes mellitus do tipo 1 podem sugerir síndrome poliglandular autoimune do tipo 2, com diabetes coexistente e IAP.[9]Betterle C, Presotto F, Furmaniak J. Epidemiology, pathogenesis, and diagnosis of Addison's disease in adults. J Endocrinol Invest. 2019 Dec;42(12):1407-33.
http://www.ncbi.nlm.nih.gov/pubmed/31321757?tool=bestpractice.com
Exame físico
A maioria dos pacientes apresenta perda de peso significativa secundária à anorexia.
A característica mais distintiva da IAP é a hiperpigmentação mocosa e cutânea generalizada, que é mais acentuada em áreas de maior fricção, como palmas, articulações e cicatrizes.[3]Husebye ES, Pearce SH, Krone NP, et al. Adrenal insufficiency. Lancet. 2021 Feb 13;397(10274):613-29.
http://www.ncbi.nlm.nih.gov/pubmed/33484633?tool=bestpractice.com
Isso ocorre em decorrência de altos níveis de hormônio adrenocorticotrófico (ACTH), que estimula os receptores de melanocortina dérmica.[Figure caption and citation for the preceding image starts]: Homem jovem com hiperpigmentação generalizada e insuficiência adrenal primária: pigmentação exacerbada nos pontos de pressão dos cotovelosDo acervo de T. Joseph McKenna, MD; usado com permissão [Citation ends]. [Figure caption and citation for the preceding image starts]: Mãos de paciente com insuficiência adrenal primária mostrando: hiperpigmentação exacerbada na superfície dorsal exposta ao sol e pregas; área de vitiligo na pele, sobre a articulação metacarpofalângicaDo acervo de T. Joseph McKenna, MD; usado com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Mãos de paciente com insuficiência adrenal primária mostrando: hiperpigmentação exacerbada na superfície dorsal exposta ao sol e pregas; área de vitiligo na pele, sobre a articulação metacarpofalângicaDo acervo de T. Joseph McKenna, MD; usado com permissão [Citation ends].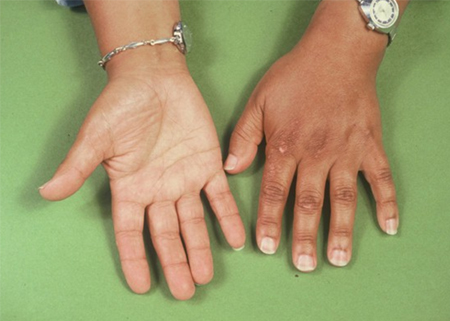 [Figure caption and citation for the preceding image starts]: Área de hiperpigmentação bucal em que houve mordida da membrana mucosa durante a mastigaçãoDo acervo de T. Joseph McKenna, MD; usado com permissão [Citation ends].
[Figure caption and citation for the preceding image starts]: Área de hiperpigmentação bucal em que houve mordida da membrana mucosa durante a mastigaçãoDo acervo de T. Joseph McKenna, MD; usado com permissão [Citation ends].
A hipotensão postural causada por deficiência de mineralocorticoides é comum.[2]Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2016 Feb;101(2):364-89.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4880116
http://www.ncbi.nlm.nih.gov/pubmed/26760044?tool=bestpractice.com
[9]Betterle C, Presotto F, Furmaniak J. Epidemiology, pathogenesis, and diagnosis of Addison's disease in adults. J Endocrinol Invest. 2019 Dec;42(12):1407-33.
http://www.ncbi.nlm.nih.gov/pubmed/31321757?tool=bestpractice.com
Em mulheres na pré-menopausa, a perda de androgênios adrenais resulta em perda significativa de pelos pubianos e axilares.[2]Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2016 Feb;101(2):364-89.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4880116
http://www.ncbi.nlm.nih.gov/pubmed/26760044?tool=bestpractice.com
[9]Betterle C, Presotto F, Furmaniak J. Epidemiology, pathogenesis, and diagnosis of Addison's disease in adults. J Endocrinol Invest. 2019 Dec;42(12):1407-33.
http://www.ncbi.nlm.nih.gov/pubmed/31321757?tool=bestpractice.com
Outros sinais físicos de autoimunidade, como vitiligo, embranquecimento prematuro do cabelo e bócio por tireoidite de Hashimoto podem estar presentes.
Avaliação laboratorial
Em uma situação de emergência, o médico não deve esperar pelos resultados dos exames de sangue para administrar o tratamento. Em pacientes com doença aguda e sintomas ou sinais inexplicados, sugestivos de insuficiência adrenal, o sangue é coletado para nível de cortisol e ACTH basal, e o tratamento é administrado imediatamente.[2]Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2016 Feb;101(2):364-89.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4880116
http://www.ncbi.nlm.nih.gov/pubmed/26760044?tool=bestpractice.com
Investigações adicionais são apropriadas em uma situação de menos urgência para confirmar a insuficiência adrenal e determinar a etiologia.[2]Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2016 Feb;101(2):364-89.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4880116
http://www.ncbi.nlm.nih.gov/pubmed/26760044?tool=bestpractice.com
Além da medição do nível de cortisol sérico, as investigações focam na integridade do principal peptídeo estimulante do córtex adrenal (ACTH plasmático), bem como nas classes de hormônios secretados pelo córtex adrenal: glicocorticoides (cortisol); mineralcorticoides (aldosterona); e androgênios adrenais (desidroepiandrosterona [DHEA], bem como seu conjugado sulfatado DHEA-S).[3]Husebye ES, Pearce SH, Krone NP, et al. Adrenal insufficiency. Lancet. 2021 Feb 13;397(10274):613-29.
http://www.ncbi.nlm.nih.gov/pubmed/33484633?tool=bestpractice.com
É esperado que, com a IAP, haja perda de secreção de cortisol, aldosterona e DHEA/DHEA-S pelo córtex adrenal e que isso esteja associado a um aumento compensatório no ACTH plasmático. Por outro lado, os pacientes com insuficiência adrenal central (ou seja, uma causa secundária ou terciária), terão menor secreção de ACTH e, consequentemente, apresentarão redução na secreção de cortisol e DHEA/DHEA-S, enquanto a função de mineralcorticoides (aldosterona) permanece relativamente intacta.[4]Hahner S, Ross RJ, Arlt W, et al. Adrenal insufficiency. Nat Rev Dis Primers. 2021 Mar 11;7(1):19.
http://www.ncbi.nlm.nih.gov/pubmed/33707469?tool=bestpractice.com
Medidas dos níveis de cortisol e ACTH
Cortisol
O primeiro teste diagnóstico é a medição do nível de cortisol sérico, de preferência pela manhã, quando espera-se que esteja em seu nível mais alto.[27]Karaca Z, Grossman A, Kelestimur F. Investigation of the hypothalamo-pituitary-adrenal (HPA) axis: a contemporary synthesis. Rev Endocr Metab Disord. 2021 Jun;22(2):179-204.
http://www.ncbi.nlm.nih.gov/pubmed/33770352?tool=bestpractice.com
Um nível de cortisol sérico aleatório também é aceitável, desde que seja interpretado no contexto da atividade do paciente e seu nível de estresse. Isso pode ser usado como um teste de exclusão.
Os valores de cortisol variam de acordo com o ensaio usado, e o uso de faixas de referência proporcionadas pelo laboratório correspondente é recomendado.[4]Hahner S, Ross RJ, Arlt W, et al. Adrenal insufficiency. Nat Rev Dis Primers. 2021 Mar 11;7(1):19.
http://www.ncbi.nlm.nih.gov/pubmed/33707469?tool=bestpractice.com
O cortisol sérico é ligado 80% à globulina de ligação ao cortisol (CBG) e 10% a 15% à albumina; assim, distúrbios que afetam os níveis de CBG ou albumina influenciam nos níveis de cortisol.[2]Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2016 Feb;101(2):364-89.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4880116
http://www.ncbi.nlm.nih.gov/pubmed/26760044?tool=bestpractice.com
Os níveis de CBG são afetados pelos níveis de estrogênio, por algumas doenças (por exemplo, doença hepática ou síndrome nefrótica) e por algumas mutações hereditárias.[8]Barthel A, Benker G, Berens K, et al. An update on Addison's disease. Exp Clin Endocrinol Diabetes. 2019 Feb;127(2-03):165-75.
https://www.doi.org/10.1055/a-0804-2715
http://www.ncbi.nlm.nih.gov/pubmed/30562824?tool=bestpractice.com
Um nível abaixo de 140 nanomoles/L (5 microgramas/dL) é altamente sugestivo de insuficiência adrenal.[2]Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2016 Feb;101(2):364-89.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4880116
http://www.ncbi.nlm.nih.gov/pubmed/26760044?tool=bestpractice.com
Um valor de corte mais baixo de 85 nanomoles/L (3 microgramas/dL) pode reduzir o número de pacientes que precisam de testes de estímulo.[27]Karaca Z, Grossman A, Kelestimur F. Investigation of the hypothalamo-pituitary-adrenal (HPA) axis: a contemporary synthesis. Rev Endocr Metab Disord. 2021 Jun;22(2):179-204.
http://www.ncbi.nlm.nih.gov/pubmed/33770352?tool=bestpractice.com
Um nível de cortisol acima de 500 nanomoles/L (18 microgramas/dL) efetivamente descarta o diagnóstico de insuficiência adrenal.[2]Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2016 Feb;101(2):364-89.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4880116
http://www.ncbi.nlm.nih.gov/pubmed/26760044?tool=bestpractice.com
[28]Kazlauskaite R, Evans AT, Villabona CV, et al. Corticotropin tests for hypothalamic-pituitary-adrenal insufficiency: a metaanalysis. J Clin Endocrinol Metab. 2008 Nov;93(11):4245-53.
https://academic.oup.com/jcem/article/93/11/4245/2627237
http://www.ncbi.nlm.nih.gov/pubmed/18697868?tool=bestpractice.com
Os pacientes com suspeita de insuficiência adrenal e níveis de cortisol de 140 nanomoles/L a 500 nanomoles/L (5 a 18 microgramas/dL) requerem exames adicionais com um teste de estímulo com o hormônio adrenocorticotrófico (ACTH) em altas doses para confirmar o funcionamento adrenal.[4]Hahner S, Ross RJ, Arlt W, et al. Adrenal insufficiency. Nat Rev Dis Primers. 2021 Mar 11;7(1):19.
http://www.ncbi.nlm.nih.gov/pubmed/33707469?tool=bestpractice.com
Hormônio adrenocorticotrópico (ACTH)
O nível de ACTH plasmático deve ser determinado em conjunto com o nível de cortisol sérico.
A presença de um nível baixo de cortisol sérico com um nível elevado de ACTH plasmático (duas vezes o limite superior do intervalo de referência de ACTH plasmático) indica IAP.[3]Husebye ES, Pearce SH, Krone NP, et al. Adrenal insufficiency. Lancet. 2021 Feb 13;397(10274):613-29.
http://www.ncbi.nlm.nih.gov/pubmed/33484633?tool=bestpractice.com
[9]Betterle C, Presotto F, Furmaniak J. Epidemiology, pathogenesis, and diagnosis of Addison's disease in adults. J Endocrinol Invest. 2019 Dec;42(12):1407-33.
http://www.ncbi.nlm.nih.gov/pubmed/31321757?tool=bestpractice.com
Às vezes, um conjunto bioquímico similar de dados pode ser observado em pacientes que se recuperam da supressão crônica do eixo hipotálamo-hipófise-adrenal (HHA).
Os pacientes com insuficiência adrenal central terão níveis baixos ou baixos-normais de ACTH em conjunto com valores baixos/baixos a normais de cortisol.[9]Betterle C, Presotto F, Furmaniak J. Epidemiology, pathogenesis, and diagnosis of Addison's disease in adults. J Endocrinol Invest. 2019 Dec;42(12):1407-33.
http://www.ncbi.nlm.nih.gov/pubmed/31321757?tool=bestpractice.com
teste de estímulo com o hormônio adrenocorticotrópico (ACTH)
Os valores ideais de cortisol devem ser individualizados, dependendo do ensaio usado, pois ensaios mais novos (por exemplo, imunoensaios de anticorpos monoclonais ou espectrometria de massa) podem ter um limiar mais baixo e resultar em teste falso-positivo.[29]Javorsky BR, Raff H, Carroll TB, et al. New cutoffs for the biochemical diagnosis of adrenal insufficiency after ACTH stimulation using specific cortisol assays. J Endocr Soc. 2021 Apr 1;5(4):bvab022.
https://www.doi.org/10.1210/jendso/bvab022
http://www.ncbi.nlm.nih.gov/pubmed/33768189?tool=bestpractice.com
O valor de 500 nanomoles/L (18 microgramas/dL) deriva de ensaios de anticorpos policlonais. Os valores de corte também precisam ser individualizados de acordo com o tipo de teste de estímulo com o hormônio adrenocorticotrófico (ACTH) usado.[30]Cho HY, Kim JH, Kim SW, et al. Different cut-off values of the insulin tolerance test, the high-dose short Synacthen test (250 μg) and the low-dose short Synacthen test (1 μg) in assessing central adrenal insufficiency. Clin Endocrinol (Oxf). 2014 Jul;81(1):77-84.
http://www.ncbi.nlm.nih.gov/pubmed/24382108?tool=bestpractice.com
Se níveis de cortisol abaixo de 500 nanomoles/L (18 microgramas/dL) forem encontrados em 30 ou 60 minutos após a estimulação por ACTH, o diagnóstico de insuficiência adrenal é altamente provável.[2]Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2016 Feb;101(2):364-89.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4880116
http://www.ncbi.nlm.nih.gov/pubmed/26760044?tool=bestpractice.com
[4]Hahner S, Ross RJ, Arlt W, et al. Adrenal insufficiency. Nat Rev Dis Primers. 2021 Mar 11;7(1):19.
http://www.ncbi.nlm.nih.gov/pubmed/33707469?tool=bestpractice.com
Níveis de cortisol acima de 500 nanomoles/L (18 microgramas/dL) 30 ou 60 minutos após a administração de uma dose elevada de ACTH (250 microgramas) descartam o diagnóstico de insuficiência adrenal primária (IAP) na maioria dos casos.[8]Barthel A, Benker G, Berens K, et al. An update on Addison's disease. Exp Clin Endocrinol Diabetes. 2019 Feb;127(2-03):165-75.
https://www.doi.org/10.1055/a-0804-2715
http://www.ncbi.nlm.nih.gov/pubmed/30562824?tool=bestpractice.com
No entanto, alguns pacientes com IAP leve ou precoce ou insuficiência adrenal central parcial manifestam um aumento adequado nos níveis de cortisol após um teste de estímulo com o hormônio adrenocorticotrófico (ACTH) em doses altas.[27]Karaca Z, Grossman A, Kelestimur F. Investigation of the hypothalamo-pituitary-adrenal (HPA) axis: a contemporary synthesis. Rev Endocr Metab Disord. 2021 Jun;22(2):179-204.
http://www.ncbi.nlm.nih.gov/pubmed/33770352?tool=bestpractice.com
[31]Gonzalez-Gonzalez JG, De la Garza-Hernandez NE, Mancillas-Adame LG, et al. A high-sensitivity test in the assessment of adrenocortical insufficiency: 10 microg vs 250 microg cosyntropin dose assessment of adrenocortical insufficiency. J Endocrinol. 1998 Nov;159(2):275-80.
https://joe.bioscientifica.com/view/journals/joe/159/2/275.xml
http://www.ncbi.nlm.nih.gov/pubmed/9795368?tool=bestpractice.com
Nas situações em que houver essa suspeita, os pacientes devem repetir o teste de estímulo usando uma dose baixa de ACTH (1 micrograma).[28]Kazlauskaite R, Evans AT, Villabona CV, et al. Corticotropin tests for hypothalamic-pituitary-adrenal insufficiency: a metaanalysis. J Clin Endocrinol Metab. 2008 Nov;93(11):4245-53.
https://academic.oup.com/jcem/article/93/11/4245/2627237
http://www.ncbi.nlm.nih.gov/pubmed/18697868?tool=bestpractice.com
Uma metanálise concluiu que os testes de estímulo com o hormônio adrenocorticotrófico em dose alta e baixa são satisfatórios para indicar a presença de insuficiência adrenal secundária, mas os dados sobre a IAP não são suficientes para estimar a precisão diagnóstica, de forma que nenhum foi confiável para codificar consistentemente o distúrbio.[32]Ospina NS, Al Nofal A, Bancos I, et al. ACTH stimulation tests for the diagnosis of adrenal insufficiency: systematic review and meta-analysis. J Clin Endocrinol Metab. 2016 Feb;101(2):427-34.
https://academic.oup.com/jcem/article/101/2/427/2810551
http://www.ncbi.nlm.nih.gov/pubmed/26649617?tool=bestpractice.com
Pacientes com teste de estímulo com o ACTH anormal compatível com insuficiência adrenal necessitam de avaliação laboratorial adicional para determinar o tipo de insuficiência adrenal.
O nível de ACTH plasmático já deve ter sido medido. Se estiver alto, a causa é a IAP. Um valor baixo ou baixo-normal indicaria um diagnóstico de insuficiência adrenal secundária.[4]Hahner S, Ross RJ, Arlt W, et al. Adrenal insufficiency. Nat Rev Dis Primers. 2021 Mar 11;7(1):19.
http://www.ncbi.nlm.nih.gov/pubmed/33707469?tool=bestpractice.com
[9]Betterle C, Presotto F, Furmaniak J. Epidemiology, pathogenesis, and diagnosis of Addison's disease in adults. J Endocrinol Invest. 2019 Dec;42(12):1407-33.
http://www.ncbi.nlm.nih.gov/pubmed/31321757?tool=bestpractice.com
As medições de atividade de renina plasmática e níveis de aldosterona sérica são importantes quando há suspeita de IAP.[2]Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2016 Feb;101(2):364-89.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4880116
http://www.ncbi.nlm.nih.gov/pubmed/26760044?tool=bestpractice.com
Em pacientes com IAP, todo o córtex adrenal é envolvido e a biossíntese da aldosterona, bem como a do cortisol, é comprometida. Como compensação, os níveis de renina elevam-se, mas de forma ineficaz. Um nível baixo de aldosterona plasmática associado com uma atividade de renina plasmática alta medida simultaneamente indica IAP.[2]Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2016 Feb;101(2):364-89.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4880116
http://www.ncbi.nlm.nih.gov/pubmed/26760044?tool=bestpractice.com
[4]Hahner S, Ross RJ, Arlt W, et al. Adrenal insufficiency. Nat Rev Dis Primers. 2021 Mar 11;7(1):19.
http://www.ncbi.nlm.nih.gov/pubmed/33707469?tool=bestpractice.com
Em contraste, na insuficiência adrenal secundária, o eixo renina-angiotensina-aldosterona está intacto, embora discretamente menos responsivo, como uma consequência das concentrações de cortisol suprimidas.
A medição dos níveis de DHEA/DHEA-S é útil na avaliação da insuficiência adrenal, pois esses androgênios adrenais são os primeiros a serem perdidos quando a função do córtex adrenal é comprometida.[2]Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2016 Feb;101(2):364-89.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4880116
http://www.ncbi.nlm.nih.gov/pubmed/26760044?tool=bestpractice.com
[27]Karaca Z, Grossman A, Kelestimur F. Investigation of the hypothalamo-pituitary-adrenal (HPA) axis: a contemporary synthesis. Rev Endocr Metab Disord. 2021 Jun;22(2):179-204.
http://www.ncbi.nlm.nih.gov/pubmed/33770352?tool=bestpractice.com
Níveis abaixo do limite inferior do normal para a idade e o sexo são um sinal inicial útil de IAP, mas não podem ser usados de maneira isolada para fazer o diagnóstico, pois os níveis podem estar baixos, principalmente em indivíduos com idade avançada, sem IAP.[2]Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2016 Feb;101(2):364-89.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4880116
http://www.ncbi.nlm.nih.gov/pubmed/26760044?tool=bestpractice.com
Como a secreção de DHEA/DHEA-S depende do ACTH, seus níveis também estarão baixos na insuficiência adrenal central. Assim, um nível baixo de DHEA-S pode indicar insuficiência adrenal, mas não identificará se é de origem primária ou central. Por outro lado, um nível normal de DHEA-S sérico torna o diagnóstico de insuficiência adrenal (primária ou central) muito improvável.[33]Nasrallah MP, Arafah BM. The value of dehydroepiandrosterone sulfate measurements in the assessment of adrenal function. J Clin Endocrinol Metab. 2003 Nov;88(11):5293-8.
https://www.doi.org/10.1210/jc.2003-030449
http://www.ncbi.nlm.nih.gov/pubmed/14602764?tool=bestpractice.com
[34]Sayyed Kassem L, El Sibai K, Chaiban J, et al. Measurements of serum DHEA and DHEA sulphate levels improve the accuracy of the low-dose cosyntropin test in the diagnosis of central adrenal insufficiency. J Clin Endocrinol Metab. 2012 Oct;97(10):3655-62.
https://www.doi.org/10.1210/jc.2012-1806
http://www.ncbi.nlm.nih.gov/pubmed/22851486?tool=bestpractice.com
Exames auxiliares
Devem ser realizados exames de sangue iniciais (hemograma completo, eletrólitos, ureia, creatinina), pois a destruição do córtex adrenal e, especialmente, a deficiência de glicocorticoides e mineralcorticoides, causam mudanças no equilíbrio hídrico e eletrolítico e alterações no hemograma.[2]Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2016 Feb;101(2):364-89.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4880116
http://www.ncbi.nlm.nih.gov/pubmed/26760044?tool=bestpractice.com
[4]Hahner S, Ross RJ, Arlt W, et al. Adrenal insufficiency. Nat Rev Dis Primers. 2021 Mar 11;7(1):19.
http://www.ncbi.nlm.nih.gov/pubmed/33707469?tool=bestpractice.com
[26]Arlt W; Society for Endocrinology Clinical Committee. Society For Endocrinology endocrine emergency guidance: emergency management of acute adrenal insufficiency (adrenal crisis) in adult patients. Endocr Connect. 2016 Sep;5(5):G1-3.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5314805
http://www.ncbi.nlm.nih.gov/pubmed/27935813?tool=bestpractice.com
A adrenalite autoimune é a causa mais comum de casos de IAP em adultos.[2]Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2016 Feb;101(2):364-89.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4880116
http://www.ncbi.nlm.nih.gov/pubmed/26760044?tool=bestpractice.com
Os anticorpos direcionados contra a enzima adrenal 21-hidroxilase estão associados à IAP autoimune, mas, apesar de sensíveis, não são necessários para o diagnóstico.[2]Bornstein SR, Allolio B, Arlt W, et al. Diagnosis and treatment of primary adrenal insufficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2016 Feb;101(2):364-89.
https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4880116
http://www.ncbi.nlm.nih.gov/pubmed/26760044?tool=bestpractice.com
[4]Hahner S, Ross RJ, Arlt W, et al. Adrenal insufficiency. Nat Rev Dis Primers. 2021 Mar 11;7(1):19.
http://www.ncbi.nlm.nih.gov/pubmed/33707469?tool=bestpractice.com
[35]Betterle C, Dal Pra C, Mantero F, et al. Autoimmune adrenal insufficiency and autoimmune polyendocrine syndromes: autoantibodies, autoantigens, and their applicability in diagnosis and disease prediction. Endocr Rev. 2002 Jun;23(3):327-64.
https://academic.oup.com/edrv/article/23/3/327/2424246
http://www.ncbi.nlm.nih.gov/pubmed/12050123?tool=bestpractice.com
Em pacientes em que a causa subjacente ainda é incerta, recomenda-se realizar exames de imagem das glândulas adrenais, por meio de tomografia computadorizada e ressonância nuclear magnética, para determinar outras causas, como infecção, hemorragia ou doença metastática.[4]Hahner S, Ross RJ, Arlt W, et al. Adrenal insufficiency. Nat Rev Dis Primers. 2021 Mar 11;7(1):19.
http://www.ncbi.nlm.nih.gov/pubmed/33707469?tool=bestpractice.com
Se houver suspeita de insuficiência adrenal secundária, mas a resposta do cortisol ao ACTH não for diagnóstica, um teste de todo o eixo hipotálamo-hipófise-adrenal (HHA) pode ser útil (por exemplo, o teste de tolerância à insulina ou o teste noturno de metirapona). O teste de tolerância à insulina mede a resposta do cortisol à hipoglicemia induzida pela insulina.[28]Kazlauskaite R, Evans AT, Villabona CV, et al. Corticotropin tests for hypothalamic-pituitary-adrenal insufficiency: a metaanalysis. J Clin Endocrinol Metab. 2008 Nov;93(11):4245-53.
https://academic.oup.com/jcem/article/93/11/4245/2627237
http://www.ncbi.nlm.nih.gov/pubmed/18697868?tool=bestpractice.com
[36]Erturk E, Jaffe CA, Barkan AL. Evaluation of the integrity of the hypothalamic-pituitary-adrenal axis by insulin hypoglycemia test. J Clin Endocrinol Metab. 1998 Jul;83(7):2350-4.
https://academic.oup.com/jcem/article/83/7/2350/2865342
http://www.ncbi.nlm.nih.gov/pubmed/9661607?tool=bestpractice.com
Vigilância é necessária em virtude de a hipoglicemia ser um desfecho. O teste é contraindicado em pacientes idosos e em pessoas com doença cardiovascular ou transtorno convulsivo. Outra opção para investigar o eixo HHA é o teste noturno da metirapona mede a resposta do 11-desoxicortisol e do cortisol à metirapona.[37]Fiad TM, Kirby JM, Cunningham SK, et al. The overnight single-dose metyrapone test is a simple and reliable index of the hypothalamic-pituitary-adrenal axis. Clin Endocrinol (Oxf). 1994 May;40(5):603-9.
http://www.ncbi.nlm.nih.gov/pubmed/8013141?tool=bestpractice.com
Infelizmente, a disponibilidade da metirapona é limitada nos EUA; ela pode ser obtida em farmácias especializadas. Em muitos países, a metirapona está disponível por meio do fabricante.