Abordaje
La mayoría de los pacientes con retraso temporal o funcional presentan un desarrollo espontáneo de la pubertad y logran un potencial de crecimiento adecuado. Sin embargo, los pacientes con un defecto orgánico, como los que tienen una deficiencia congénita de gonadotropina o anomalías gonadales, necesitan un tratamiento temprano para lograr una estatura final óptima y características sexuales secundarias. En la ausencia de cambios puberales, la velocidad de crecimiento en estatura disminuye de 5 cm por año a la edad de 12 años a razón de 1 cm por año cada año en que no se manifiesta la pubertad a partir de esa edad. Un endocrinólogo pediátrico experimentado en el diagnóstico y tratamiento de retraso puberal debe evaluar a los pacientes.
Antecedentes
La mayoría de los pacientes que presentan retraso puberal son niños. Muchos pacientes buscan ayuda médica debido a un crecimiento lento más que a un desarrollo puberal lento, así como debido a la preocupación de que la pubertad no haya comenzado.
La evaluación debe incluir inicialmente antecedentes detallados de las causas de un retraso temporal. Cualquier enfermedad crónica puede llevar al retraso puberal con o sin estatura baja y aumento de peso insuficiente. Las afecciones frecuentes incluyen desnutrición, cardiopatía crónica, asma moderada a grave, fibrosis quística, enfermedad celíaca, enfermedad inflamatoria del intestino (enfermedad de Crohn y colitis ulcerosa), enfermedades inflamatorias (p. ej. la artritis idiopática juvenil), insuficiencia renal crónica, neoplasia maligna crónica y diabetes mellitus mal controlada. Se debe buscar antecedentes de patrones de alimentación anormales o pérdida de peso reciente en las niñas. Algunas veces, puede asociarse con rutinas de ejercicio intenso y se debe sospechar de un trastorno de la alimentación como anorexia nerviosa o bulimia nerviosa.
El retraso constitucional, también conocido como retraso constitucional del crecimiento y la pubertad (RCCP), pubertad retrasada autolimitada o pubertad retrasada aislada, es la causa más común de retraso puberal, tanto en hombres como en mujeres.[1][20][37] Suele darse en familias y está asociada a la baja estatura. Deben registrarse los antecedentes familiares, incluidas las estaturas de ambos progenitores y sus edades al inicio del desarrollo puberal, así como la edad materna de la menarquia.[37]
Los antecedentes pueden revelar cirugía previa o radioterapia al cerebro (que puede producir deficiencia de gonadotropinas) o al abdomen o pelvis (daño gonadal) debido a neoplasia maligna previa. Varios agentes quimioterapéuticos pueden producir retraso puberal. El dolor testicular o infecciones virales recientes como paperas pueden indicar infección o torsión testicular. La histiocitosis, la anemia falciforme y la sobrecarga de hierro (asociada con transfusiones) pueden producir deficiencia de gonadotropina permanente.
Es posible que se presenten síntomas que sugieren otros déficits hormonales como un retraso del crecimiento por deficiencia de la hormona de crecimiento o síntomas de hipotiroidismo, lo que puede indicar una causa central (hipotalámico-hipofisaria). Esto se puede asociar a la pérdida del olfato (anosmia), lo que sugiere síndrome de Kallman.
Alteraciones de múltiples glándulas endocrinas como retraso puberal, además de diabetes mellitus y/o enfermedad de Addison, deben sugerir un trastorno autoinmune. En raras ocasiones, la deficiencia genética congénita de gonadotrofinas puede asociarse a una hipoplasia suprarrenal congénita, y los antecedentes pueden ser indicativos de pérdida de sal o crisis suprarrenal. Puede haberse llegado ya al diagnóstico, en cuyo caso el paciente puede estar recibiendo tratamiento con glucocorticoides y mineralocorticoides.[Figure caption and citation for the preceding image starts]: Crecimiento en un paciente con retraso constitucional del crecimiento y la pubertad; la X indica la edad ósea; la flecha indica la edad de inicio del desarrollo puberalDe la colección del Dr. A. Mehta [Citation ends].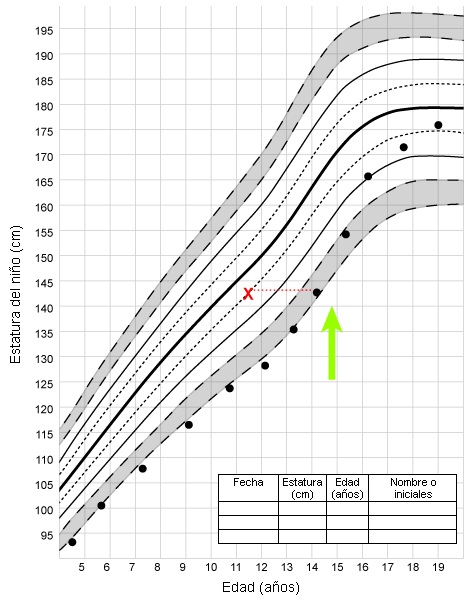
Etapas de Tanner
El desarrollo puberal y su evolución se evalúan mejor con las etapas de Tanner.[38][39]
El primer signo de pubertad en los niños es el aumento en el tamaño de los testículos. Esto es seguido por cambios en el pene y el escroto. El tamaño testicular se documenta a través de la medición del eje más largo o por el volumen testicular utilizando el orquidómetro de Prader.
[Figure caption and citation for the preceding image starts]: Orquidómetro de PraderCreado por BMJ Knowledge Centre [Citation ends].

[Figure caption and citation for the preceding image starts]: Método para comparar el tamaño testicular utilizando el orquidómetro de PraderDe la colección del Dr. A. Mehta [Citation ends].
 Un volumen de 4 mL o una longitud de 2.5 cm define el inicio de la pubertad.[39] El crecimiento de vello axilar y otros cambios como los cambios en la voz y un aumento en la velocidad de crecimiento se producen en la etapa media y final de la pubertad. El vello facial no aparece hasta la etapa final de la pubertad.
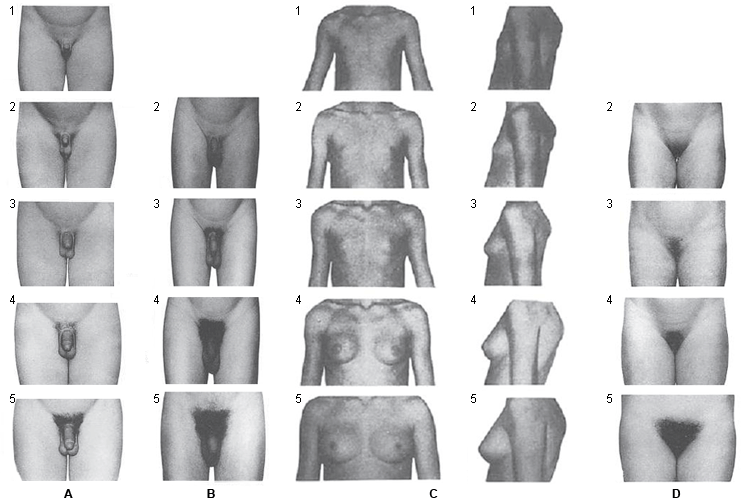
Un volumen de 4 mL o una longitud de 2.5 cm define el inicio de la pubertad.[39] El crecimiento de vello axilar y otros cambios como los cambios en la voz y un aumento en la velocidad de crecimiento se producen en la etapa media y final de la pubertad. El vello facial no aparece hasta la etapa final de la pubertad.El primer signo demostrable de pubertad en niñas es el desarrollo mamario. Se debe tener precaución al examinar el tejido mamario en niñas obesas, ya que la grasa puede confundirse con tejido mamario. El vello púbico y axilar, el acné y el olor corporal se desarrollan como resultado de los andrógenos secretados por la glándula suprarrenal. El pico de crecimiento se produce en el etapa 3 de Tanner de desarrollo mamario y la menarquia se produce normalmente junto con la etapa 4 de Tanner de desarrollo mamario.[Figure caption and citation for the preceding image starts]: Etapas de Tanner: A, estándares de evaluación genital en niños; B, estándares de evaluación del vello púbico en niños; C, estándares de evaluación de los senos en niñas; D, estándares de evaluación del vello púbico en niñasAdaptado de Marshall WA, Tanner JM. Arch Dis Child. 1970;45:13-23; Marshall WA, Tanner JM. Arch Dis Child. 1969;44:291-303 [Citation ends].
Exploración física
Se deben registrar la estatura y el peso actuales y previos de manera precisa. La mayoría de los pacientes que presentan retraso puberal son niños con retraso constitucional que tienen un examen normal, además de estatura baja y una velocidad de crecimiento ligeramente inferior a la media para su edad y sexo. Los que tienen un retraso temporal, como ocurre en las enfermedades crónicas, pueden tener las alteraciones típicas de ese trastorno. Es importante evaluar al paciente en busca de rasgos de desnutrición.
Se debe evaluar a los pacientes en busca de desproporción corporal. Los niños que tienen el síndrome de Klinefelter son de estatura alta y mayor longitud de las extremidades inferiores. Además, pueden presentar testículos disgénicos (pequeños y blandos a la palpación) y retraso en el desarrollo, y pueden tener ginecomastia.[27]
Las niñas con síndrome de Turner suelen ser de baja estatura y presentan varios rasgos dismórficos, como entradas bajas, cuello palmeado y orejas prominentes rotadas hacia atrás. Las características al momento de la presentación en muchos casos pueden ser más sutiles y deben realizarse investigaciones para descartar el síndrome de Turner en las niñas que presentan un retraso puberal.[40][41] Consultar Síndrome de Turner.
Pueden existir otros rasgos que sugieran un diagnóstico sindrómico (p. ej., Prader-Willi, Bardet-Biedl, CHARGE, o displasia septoóptica).
Debe realizarse un examen detallado del sistema nervioso central en busca de cualquier déficit focal que sugiera un tumor intracraneal. Debe comprobarse el sentido del olfato; la anosmia sugiere el síndrome de Kallman, el cual se asocia con el hipogonadismo hipogonadotrópico orgánico y nervios olfativos hipoplásicos.[21] Algunos pacientes con síndrome de Kallmann tienen un defecto genético en genes como ANOS1, FGF8, FGFR1, PROK2 o PROKR2.
[Figure caption and citation for the preceding image starts]: Evaluación de una paciente con retraso puberal. RCCP: retraso constitucional del crecimiento y la pubertad; PR: pubertad retrasada; FT4: T4 libre; GHD: deficiencia de hormona de crecimiento; PRL: prolactina; PFT: prueba de función tiroideaEndocr Rev. 2019 Oct; 40(5): 1285–1317; usado con autorización [Citation ends].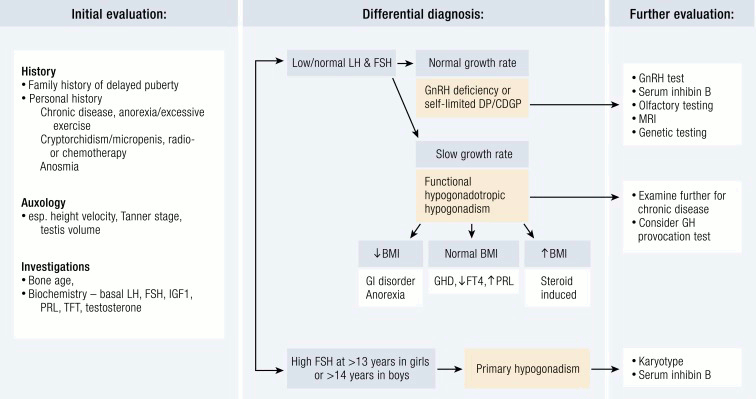
Pruebas iniciales
Las pruebas iniciales deben incluir una radiografía de la muñeca no dominante (normalmente, la izquierda) para estimar la edad ósea. La edad ósea indica si existe o no retraso constitucional y ayuda a predecir el intervalo estimado de estatura adulta y su relación con la estatura media parental.[1] Se compara la aparición de centros epifisarios representativos en la radiografía con los estándares publicados adecuados para la edad y el sexo. El método más frecuentemente utilizado es el de Greulich y Pyle.[42]
La medición en el laboratorio de las concentraciones de la hormona foliculoestimulante (FSH) la hormona luteinizante (LH) séricas ayudará a diferenciar a pacientes con hipogonadismo hipogonadotrópico (niveles bajos) e hipogonadismo hipergonadotrópico (niveles elevados).[1] Se deben realizar las mediciones en una muestra de sangre temprano por la mañana utilizando un ensayo pediátrico ultrasensible. Como el eje hipotalámico-hipofisario-gonadal está en reposo hasta aproximadamente los 10 a 12 años de edad, los niveles de esas hormonas en niños <12 años de edad deben interpretarse con precaución. Puede ser difícil distinguir el retraso constitucional de la deficiencia orgánica de gonadotrofinas, ya que las concentraciones basales de gonadotrofinas pueden ser bajas o normales en ambos grupos.
Las investigaciones adicionales y el tratamiento dependen de los antecedentes, la gravedad del retraso, la estatura y los resultados de la medición de gonadotropina basal.
Pruebas posteriores
Prueba de hormona liberadora de gonadotropinas (GnRH)
Puede considerarse en todos los pacientes con gonadotropinas basales bajas; sin embargo, la sensibilidad y la especificidad son limitadas.[43] Las niñas con el síndrome de Turner y otros pacientes con anomalías gonadales, generalmente presentan marcados aumentos en las concentraciones basales de FSH y LH séricas (en el síndrome de Turner entre 8 y 9 años de edad) y generalmente no se indica la prueba de GnRH en estos pacientes.[44]
La hormona liberadora de la hormona luteinizante (LHRH) se utiliza para estimular las gonadotropinas. Se ha estudiado ampliamente en pacientes con gonadotropinas basales bajas, pero no siempre ayuda a diferenciar entre el retraso constitucional y la deficiencia orgánica de gonadotropinas, ya que las concentraciones de FSH sérica estimulada y LH tras la hormona liberadora de la hormona luteinizante (LHRH) pueden ser bajas en ambos grupos, o pueden ser normales y, por tanto, falsamente tranquilizadoras en un individuo con deficiencia hipotalámica de la hormona liberadora de gonadotropina (GnRH) pero función hipofisaria preservada.[45] Por consiguiente, no permite aclarar si un individuo avanzará espontáneamente para la pubertad o tiene un defecto permanente. Por lo tanto, algunos pacientes con un retraso temporal pueden necesitar tratamiento con esteroides sexuales para inducir la pubertad, pero cuando se les vuelve a evaluar, una vez completado el crecimiento y la pubertad, con una prueba de suspensión del tratamiento, pueden revelar concentraciones séricas normales de FSH y LH.
Inhibina B y hormona antimülleriana
La inhibina B basal es una investigación bioquímica prometedora para el diagnóstico del retraso puberal. En los varones, es producida exclusivamente por las células de Sertoli de los testículos, y en las mujeres por las células de la granulosa ovárica y la placenta. Un nivel bajo de inhibina B inferior a 35 pg/mL en hombres prepúberos es un marcador sensible para discriminar el hipogonadismo hipogonadotrófico permanente del retraso puberal constitucional o autolimitado.[46] En los varones, el trío de volumen testicular pequeño (punto de corte: 1.1 ml), LH estimulada máxima baja en la prueba de estimulación con GnRH (punto de corte: 4.3 UI/l) y concentración basal baja de inhibina B (punto de corte: 61 pg/mL) se ha propuesto como el discriminador más eficaz entre estas dos condiciones.[18] En los hombres con hipogonadismo hipergonadotrófico, un nivel bajo de inhibina B en suero refleja un fallo primario de las células germinales. La utilidad de la inhibina B se ha demostrado con menos claridad en las niñas.
La hormona antimulleriana (AMH) se produce en los hombres a partir de las células de Sertoli testiculares desde el momento de la diferenciación testicular hasta la pubertad. En las mujeres, es secretado por las células de la granulosa ovárica desde el nacimiento hasta la menopausia, aunque las concentraciones en las niñas son significativamente más bajas. Los pacientes con gónadas disgenéticas tienen un nivel bajo de AMH en suero. La hormona antimulleriana (AMH) y la inhibina B indetectables son características de la anorquia congénita, pero también pueden observarse en hombres y mujeres con hipogonadismo hipogonadotrófico grave. La AMH y la inhibina B pueden utilizarse, por tanto, durante la evaluación de la función y la capacidad testicular u ovárica (como marcador del potencial de fertilidad), y como herramienta diagnóstica junto con las gonadotropinas séricas para el hipogonadismo tanto hipo como hipergonadotrófico.[47]
Prueba de estimulación con gonadotropina coriónica humana (hCG)
La prueba de hCG se utiliza para estimular la producción testicular de testosterona y se ha sugerido que es muy útil en algunos pacientes cuando se combina con la prueba de LHRH para ayudar a distinguir el retraso constitucional del hipogonadismo hipogonadotrópico.[45]
Se observa un aumento en la concentración sérica de testosterona en el retraso constitucional; se observa una disminución en la respuesta a la testosterona en el caso de hipogonadismo hipogonadotrópico.
Toma de muestras de gonadotropinas durante la noche
Pueden demostrar la pulsatilidad pero su rol en el pronóstico del retraso a largo plazo no está claro. Se realiza principalmente en un entorno de investigación.
Otras pruebas
Esteroides sexuales: la medición de los esteroides sexuales es útil solo en el contexto de las pruebas de estimulación con hCG. La medición basal de los esteroides sexuales séricos (testosterona y estradiol) rara vez es diagnóstica.
Análisis cromosómico: puede revelar el síndrome de Klinefelter (47XXY) en niños o el síndrome de Turner (45X) en niñas. El diagnóstico de síndrome de Turner (45X) debe considerarse en todas las niñas de baja estatura, con o sin otras características fenotípicas que sugieran la presencia del síndrome de Turner.[41] Según el mosaicismo, la disgenesia gonadal mixta 45X/46XY puede presentarse con genitales ambiguos en el nacimiento o con retraso puberal en la adolescencia.[40] El microarray cromosómico puede estar indicado en pacientes con hipogonadismo hipergonadotrófico con anomalías congénitas adicionales o rasgos sindrómicos.[48]
Secuenciación genética: para las personas con sospecha de hipogonadismo hipogonadotrófico congénito o síndrome de Kallmann, las pruebas de panel de secuenciación de nueva generación ya están disponibles en el Reino Unido y en algunas otras regiones del mundo.[49]
Diagnóstico mediante estudios por imágenes: debe considerarse la realización de una ecografía pélvica en niñas con síndrome de Turner u otras formas de disgenesia gonadal, así como en niñas con hipogonadismo hipogonadotrófico. En individuos con hipogonadismo hipergonadotrófico, puede revelar gónadas disgenéticas. En las personas con hipogonadismo hipogonadotrófico, es útil para evaluar la madurez uterina y ovárica (volumen, forma y grosor endometrial) para el diagnóstico y antes de iniciar el tratamiento, a fin de permitir el seguimiento de la respuesta a la terapia. El ultrasonido abdominal puede revelar alteraciones renales como agenesia o un riñón en herradura. La ecocardiografía en niñas con el síndrome de Turner puede presentar defectos cardíacos, incluidos coartación de la aorta y una válvula aórtica bicúspide. La resonancia magnética del cerebro debe considerarse si las gonadotropinas basales son bajas en ausencia de antecedentes familiares de retraso puberal. Es obligatorio si se detectan niveles deficientes de otras hormonas hipofisarias. La neuroimagen del cerebro ayuda a identificar anomalías hipotalámico-hipofisarias estructurales, defectos en la línea media del cerebro, hipoplasia olfativa y tumores hipofisarios.[1][50]
Se deben considerar otras pruebas de hormonas hipofisarias para descartar deficiencias combinadas de hormonas hipofisarias en pacientes con hipogonadismo hipogonadotrópico confirmado.
Medición de anticuerpos ováricos séricos para identificar un proceso autoinmune.
Las pruebas de función tiroidea y las concentraciones séricas de la prolactina son útiles para identificar pacientes con hipotiroidismo o hiperprolactinemia, ambas pueden presentarse con retraso puberal.
Para evaluar la producción de GnRH por el hipotálamo, se ha propuesto la medición de LH en respuesta a la estimulación con kisspeptina como prueba útil para identificar a los individuos con deficiencia de GnRH y, por tanto, con hipogonadismo hipogonadotrófico permanente. La kisspeptina estimula la pulsatilidad de la GnRH, favoreciendo así la secreción de LH y, en menor medida, de FSH. Un estudio clínico encontró que el aumento máximo de LH tras la administración de kisspeptina era más preciso para el diagnóstico de hombres con deficiencia de GnRH que las pruebas de estimulación de GnRH.[51] En un estudio paralelo realizado en adolescentes con retraso puberal (3 mujeres y 13 hombres), se demostró que el pico de LH tras la estimulación con kisspeptina era superior a la prueba de estimulación con GnRH para predecir la capacidad de avanzar en la pubertad.[52] Aunque son necesarias más investigaciones para delinear los parámetros de uso de la kisspeptina en la práctica clínica pediátrica, se trata de un área prometedora para el diagnóstico bioquímico de la deficiencia de GnRH en la adolescencia.
El uso de este contenido está sujeto a nuestra cláusula de exención de responsabilidad