Approach
A thorough history and complete physical and neurological examination is essential in any infant presenting with dystonia. This usually enables the physician to classify the dystonia according to clinical characteristics, which also suggest whether the cause is inherited or acquired. Further investigations may be required, for which the authors’ general approach is outlined in the following image.
[Figure caption and citation for the preceding image starts]: Infantile dystonia: assessment and genetic diagnosisCreated by BMJ Knowledge Centre [Citation ends].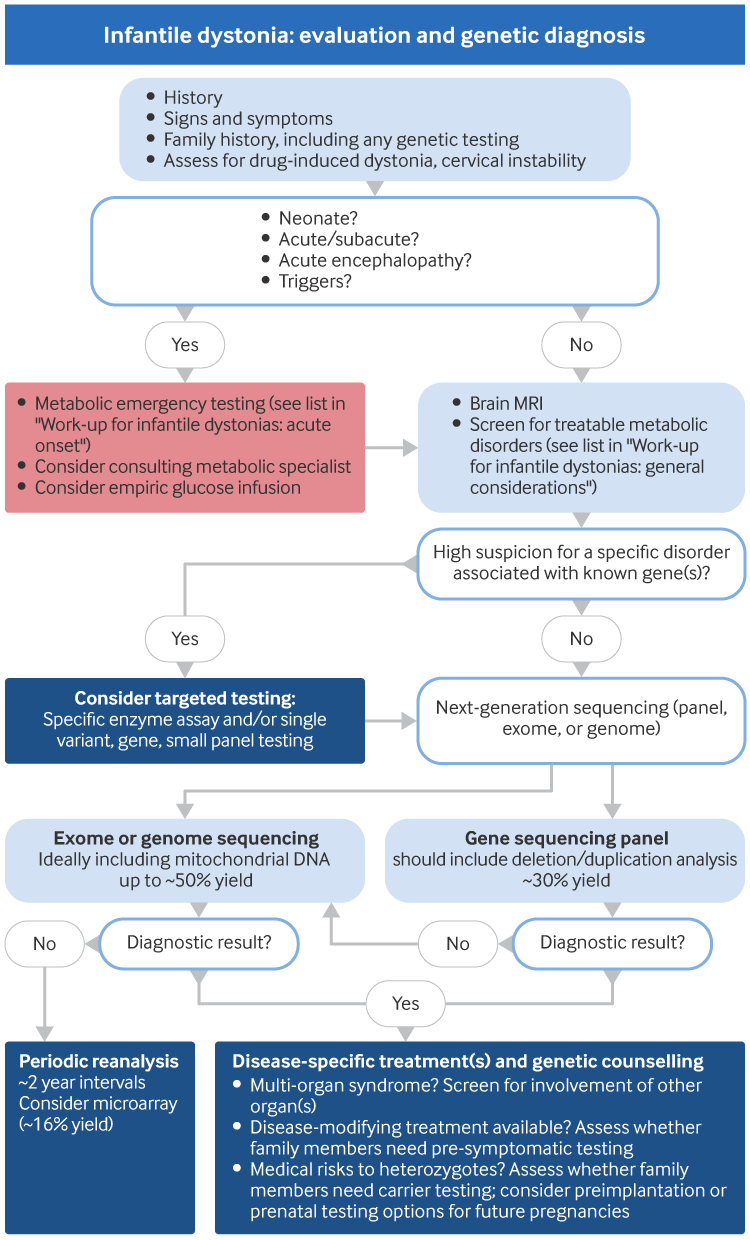
History
Essential information to be gathered from the parents includes:
Family history
Consanguinity
History of drug/toxin exposure
History of perinatal injury
History of central nervous system infection
History of head/neck trauma
Age of onset
Initial body part involved and the course of the dystonia
Precipitating factors (e.g., drugs, infections, activity, exercise); aggravating factors (e.g., fatigability) and diurnal variations; alleviating factors (e.g., sleep)
Information regarding the evolution of the dystonia (e.g., chronic, paroxysmal, or transient).[114]
Parents should be encouraged to provide photographs and to film their infants. These are extremely valuable tools in evaluating an infant with intermittent abnormal movements.
Clinical presentation
The observed clinical presentation and historical features will help to distinguish infantile dystonia from other abnormal movement disorders. It can, however, be difficult to distinguish dystonia from other causes of hypertonia in infants, such as rigidity and spasticity. For older children a quantitative assessment tool, the Hypertonia Assessment Tool, is available.[79][115] Hypertonia assessment tool Opens in new window
Dystonic movements are patterned and sustained. They repeatedly involve the same muscle groups and typically cause twisting of body parts. Dystonia usually gets worse with fatigue and emotional stress, and it gets better with sleep and relaxation.[4][116][117][118] Passive movements are resisted at very low velocity of limb movement (as distinct from spasticity, which is velocity dependent). Geste antagoniste (sensory tricks) are well described in focal dystonia affecting the neck, hand, and face. Improvement in dystonic movements is typically maintained while the trick is performed by the patients, but disappears when the alleviating gesture ends.[7]
Other differential abnormal movement disorders have the following features.
Rigidity is increased resistance to passive movements or articulations, which is uniform regardless of the speed and direction of movement (flexion or extension), traditionally compared to the resistance of a lead pipe. Resistance can be rhythmically interrupted resulting in the cogwheel phenomenon. Rigidity is most perceptible with slow movements.[5]
In spasticity, hypertonicity predominates in flexor and pronator muscles in the upper limbs, and resistance is at onset of movement and then increases with the speed of passive motion. This is the jack-knife phenomenon.
Tics are repetitive movements of skeletal or pharyngolaryngeal muscles; the latter are responsible for emission of sounds or noises. They are stereotyped, sudden, inopportune, non-propositional, absurd, irresistible movements of variable intensity. The patient can reproduce tics or control them voluntarily. Tics may persist during sleep.
Tremor is a rhythmic oscillation of a part of the body around a fixed point or place. It results from alternating synchronous contractions of antagonistic muscles reciprocally innervated. Rhythmicity is the most useful characteristic for identifying tremor.
Ataxia is a disturbance in the fine control of posture and movement. The initial and most prominent feature is usually an abnormal gait.
In inherited and idiopathic dystonias without degeneration, the dystonia and/or abnormal movements usually predominate. Each type of isolated inherited dystonia has specific findings that give important clues to the exact diagnosis. Some important characteristics that may suggest a particular dystonia syndrome lie along the clinical characteristics axis of dystonia classification. In addition to age of onset and whether there are additional motor features (isolated, combined, complex dystonias), they include the distribution of dystonia and its temporal pattern, including whether the course is static or progressive, and any variability such as diurnal variation or paroxysmal or action-induced occurrence. There is some phenotypic overlap between diseases of different dystonia genes due to variability in clinical presentation: for example, some combined dystonias can initially present with apparently isolated phenomenology.
In acquired dystonias and inherited complex dystonias, the presence of other neurological and/or systemic abnormalities suggests a variety of insults. These include vascular, infectious, neoplastic, and traumatic, as well as syndromes with underlying metabolic and degenerative diseases.[114] There is usually progressive neurological deterioration in inherited dystonia due to underlying metabolic or degenerative disease. As with inherited isolated dystonia, important clues in the history and examination may lead the physician to diagnose the specific type of dystonia.
A positive family history of dystonia may suggest an inherited aetiology. However, a negative family history does not rule this out as there are dystonias where autosomal-recessive inheritance, incomplete penetrance or variable expressivity (such as only 30% penetrance in DYT-TOR1A), or de novo pathogenic variants for autosomal-dominant conditions, are seen.
Periodic follow-up of infants with dystonia is often needed to make an accurate diagnosis.
Disorders that mimic dystonia and pitfalls in diagnosis
Primary stereotypies, paroxysmal tonic upgaze of infancy, secondary infantile torticollis (SIT), and seizure should be considered in the differential diagnosis of dystonia.
Dopa-responsive dystonia (Segawa syndrome, dystonia-parkinsonism with diurnal fluctuation) is commonly misdiagnosed as spastic diplegic or quadriplegic cerebral palsy, intractable epilepsy, or hereditary spastic paraplegia.[9][10][119] It should be considered as a possible diagnosis in any child with paroxysmal progressive hypertonia of unknown origin.
Infantile hereditary hyperekplexia should be considered when a neonate is noted to have generalised rigidity. Characteristic signs are a noise- or a nose-tap induced excessive startle response, either of which produces a brief violent flexor stiffness and breath-holding.[113]
The possibility of a genetic aetiology where timely disease-specific treatment could significantly change outcome should be taken into account. Many rare movement disorders now have known treatments that can mitigate, prevent, or even reverse symptoms, summarised by the International Parkinson’s Disease Movement Disorders Society Task Force on Rare Movement Disorders.[120] Dopa-responsive dystonia (treatable with levodopa) should be considered in any patient with unknown diagnosis and normal magnetic resonance imaging (MRI). In patients with dystonia, seizures, and encephalopathy, cerebrospinal fluid (CSF) studies of tetrahydrobiopterin and neurotransmitter metabolites may detect neurotransmitter disorders with specific treatments (e.g., levodopa, tetrahydrobiopterin, folinic acid) based on the affected enzyme. Among the potential aetiologies of acute or subacute-onset dystonia with encephalopathy are organic acidaemias with toxic metabolites (where acute and chronic metabolic management can change outcome) and biotin-thiamine-responsive basal ganglia disease. Paroxysmal dystonia with or without epilepsy may be due to glucose transporter 1 (GLUT1; SLC2A1) deficiency, treated with the ketogenic diet. Rare causes of dystonia with neurodegeneration include cerebral creatine deficiency and cerebral folate transport deficiency, which may respond to dietary treatment. Brain MRI findings may suggest manganese deposition (pallidal T1 hyperintensity) in hypermanganesaemias, treatable with chelation.
Work-up for infantile dystonias
Acute onset
If the dystonia is acute-onset or part of an acute neurological deterioration, prompt evaluation is indicated for 'metabolic emergency' as well as the other urgent situations listed above, such as drug intoxication or bilirubin encephalopathy. Clinical suspicion is particularly high in infants who have previously been well and then progressively deteriorate over a period of hours to days; with new encephalopathy or seizures; where the history includes potential triggers for metabolic decompensation such as febrile illness, vomiting, or fasting; and in the neonatal period. The goal of metabolic testing is to identify disorders with small-molecule intoxication (e.g., bilirubin, ammonia, organic acids in organic acidaemias) and treat them before they progress to brain oedema or necrosis. A guide to emergency testing is available in the Vademecum Metabolicum. Vademecum Metabolicum Opens in new window Tests include:
Arterial blood gas (ABG) and blood electrolytes (evaluate for acidosis and calculate anion gap)
Blood glucose
Serum ammonia
Serum lactate
Blood uric acid
Liver transaminases, coagulation studies
Blood creatine kinase, C-reactive protein, creatinine
Urine ketones, glucose, protein (dipstick).
Additionally, bilirubin (to evaluate for kernicterus) and urine sulfite dipstick testing for sulfite oxidase deficiency (if available) may be indicated in neonates. Blood and urine samples may also be collected for additional metabolic testing such as urine organic acids as described below, because the yield is highest during the acute illness.
While the above evaluations are ongoing, empirical treatment can be considered: intravenous infusion of 10% glucose in appropriate electrolytes, with monitoring of lactate and blood pH. The purpose of infusing glucose is to reduce catabolism and thus decrease the production of the neurotoxic compound, so it is given even if the patient is not hypoglycaemic. Further management would ideally be undertaken in consultation with a metabolic specialist.
General considerations
The first priority in diagnostic testing is to evaluate for acquired and metabolic causes that have specific treatments. MRI of the brain and metabolic laboratory tests are often needed as part of this. Molecular genetic testing is then helpful to reach the diagnosis. For paediatric movement disorders in general, published diagnostic approaches based on expert opinion are available.[121] In the absence of specific consensus guidelines for infantile dystonia evaluation, the image above shows the authors’ general approach.
MRI of the brain should be performed in all infants and children where history and clinical presentation suggest that the aetiology could be acquired, lesional, or a degenerative disorder. In practice, even if a genetic dystonia is considered the most likely diagnosis, an MRI of the brain is usually performed to rule out secondary causes and to evaluate for patterns of lesions or atrophy. If magnetic resonance spectroscopy (MRS) is available, it may be performed to evaluate for patterns associated with some metabolic diseases, such as low creatine in cerebral creatine deficiency disorders that have specific treatments or high lactate in mitochondrial disorders. MRI is normal in inherited isolated dystonias but is often abnormal in acquired or degenerative dystonias.
In a patient with acute or subacute onset of symptoms due to intoxication aetiologies, such as organic acidaemias, MRI may show oedematous or necrotic basal ganglia lesions; an elevated lactate peak may suggest a mitochondrial disorder or biotin-thiamine-responsive basal ganglia disease. MRI in chronic disease may show atrophy, brain malformation, or more specific findings, such as iron deposition in neurodegeneration with brain iron accumulation or white matter involvement in hypomyelinating disorders. Computed tomography scan of the brain may be indicated to view calcifications.
The choice of any further investigations will be influenced by the clinical presentation and the possible secondary causes that are under consideration.
Electroencephalogram (EEG) or video-EEG monitoring should be considered if the history suggests seizure, especially for paroxysmal conditions such as transient idiopathic dystonia of infancy, benign paroxysmal torticollis of infancy, and acquired dystonias.
Metabolic testing can be considered in all infants with dystonia to evaluate for disorders with specific treatments. It is particularly important when the MRI of the brain is abnormal or the clinical features suggest a metabolic cause. It is sometimes performed at the same stage as an MRI of the brain. The following laboratory tests make up a basic metabolic work-up:
ABG analysis
Serum ammonia
Urine ketone analysis
Serum amino acid analysis
Urine organic acid screen for organic acidurias
Serum uric acid analysis for Lesch-Nyhan syndrome
Serum and CSF lactate and pyruvate testing for mitochondrial encephalomyopathies
Serum and CSF glucose for GLUT1
CSF studies of neurotransmitters.[28]
If the MRI of the brain is normal and the diagnosis has not been established by previous tests, a diagnostic therapeutic trial with levodopa should be considered. This test is warranted in every patient with early-onset dystonia, where the diagnosis has not been established.[122] Symptoms improve dramatically in the more common form of dopa-responsive dystonia (Segawa syndrome, dystonia-parkinsonism with diurnal fluctuation) compared with a slow or partial response in other types of dystonia.
Pathogenic variation in the familial hemiplegic migraine (FHM1) gene CACNA1A has been reported in a family with benign paroxysmal torticollis in father and son.[123] Testing is indicated if there is a family history of benign paroxysmal torticollis of infancy or a similar disorder such as hemiplegic migraine.
Automated brainstem responses, brainstem auditory-evoked responses, and full audiology evaluation should be performed if kernicterus or mitochondrial encephalomyopathies are the working diagnosis as a cause of dystonia.
Enzyme activity of hexosaminidase A and total hexosaminidase should be assayed in leukocytes or other tissues if infantile GM2 gangliosidosis is high on the differential diagnosis of dystonia.
Muscle biopsy can be considered in patients with dystonia with possible mitochondrial encephalomyopathies if specific mitochondrial genome and nuclear-encoded gene sequencing or deletion/duplication studies are not informative.
Molecular genetic testing is helpful, along with biochemical testing/enzymatic analysis if a specific enzyme deficiency is suspected.[124]
Molecular genetic testing
Genetic tests have reported diagnostic yields of 30% to 60% in children with dystonia, a higher rate than in dystonia in general (approximately 12% in cohorts that include adults).[2][125] Earlier onset, combined or complex dystonia, and familial occurrence all raise the likelihood of genetic diagnosis.[2][125] To achieve these high yields, it is important to choose a genetic testing strategy that takes into account the patient’s clinical scenario and the types of variants each test can detect.[126]
In most patients, the pathogenic genetic variant is a small sequence variant (a change of a single nucleotide or insertion/deletion of a few nucleotides) detectable by sequencing. A minority of patients have copy number variants (CNVs) such as deletions or duplications; these may involve one or more exons within the gene (intragenic CNVs), the entire gene, or multiple genes (microdeletions/duplications).
The commonly requested genetic tests are as follows.
Variant-specific tests and single-gene tests: specific tests are available for some common variants, such as a specific deletion of 3 nucleotides in many patients with DYT-TOR1A. Because these tests are designed to only detect specific variants, negative results do not rule out other variants in the same gene. If a single gene needs to be evaluated, this can be done using a combination of Sanger sequencing to identify small sequence variants and exon-level array to identify intragenic CNVs.
Gene panels: these assess a subset of genes associated with dystonia, often via next-generation sequencing (NGS) methods. Each laboratory selects which genes to analyse. Gene panels almost never cover all of the relevant genes, as dystonia can occur in patients with syndromes like developmental and epileptic encephalopathies that are not usually thought of as movement disorders.[127] Gene panels detect small sequence variants. Ideally, they will also include detection of intragenic CNVs by methods such as exon array or additional analysis of NGS data; if so, the testing laboratory will explicitly state that deletion/duplication (del/dup) testing or CNV testing will be performed. Even with these methods, gene panels may not detect larger CNVs and microdeletions/duplications that span the whole gene. Diagnostic yield depends on the number of genes analysed; a 102-gene panel had a diagnostic rate of 28% in paediatric dystonia (34% in combined and 16% in isolated dystonia).[128]
Exome and genome sequencing: these NGS methods assess either exons and adjacent sequences (exome sequencing), or sequences from across the genome (genome sequencing). Mitochondrial DNA sequencing is not necessarily included, but often useful because the differential diagnosis can include mitochondrial DNA variants such as those causing Leigh syndrome. Both exome and genome sequencing detect small variants. They can also sometimes detect CNVs with varying degrees of success.[129] The yield of exome sequencing was 60% to 66% in small sub-cohorts of patients with infantile-onset dystonia.[2][3] More generally in paediatric-onset dystonia, yields of 34% to 53% have been reported.[2][3] Genome sequencing has a higher yield than exome sequencing in dystonia.[125] This is primarily due to better sequence data quality (allowing higher sensitivity for small variants) and better identification of CNVs (which accounted for 23% of genetic diagnoses in the referenced cohort).[125]
Chromosomal microarray: these tests detect primarily large CNVs such as microdeletions/duplications. They do not detect small sequence variants. The smallest size of CNV detectable (resolution) varies between tests but is usually not sufficient to detect intragenic CNVs. In one study, 16% of patients with paediatric dyskinesia had microdeletions of known dyskinesia genes large enough to detect on microarray, with another 12% having possibly relevant microdeletions on microarray.[130]
There is no single correct choice of which test to order first, but the decision can be guided by the breadth of the differential diagnosis. Because the phenotypic spectra of many dystonia genes overlap, it is often appropriate to undertake an NGS test evaluating multiple genes – gene panel, exome, or genome sequencing – as the initial genetic test. The following considerations apply.
In general, exome or genome sequencing will offer the highest diagnostic rates, especially when the differential diagnosis is broad. It is appropriate to request exome or genome sequencing as initial testing. A gene panel does not need to be obtained first.[131] If exome or genome sequencing is ordered, the laboratory should be sent detailed clinical information including neurological features, non-neurological features, family history, any previous genetic testing, and any specific disorders/genes that are high on the differential diagnosis. The genetic testing laboratory can use this information to improve the quality of data interpretation.
When there is high suspicion for a specific disorder, single-gene or gene panel testing may have advantages over exome/genome sequencing. Besides higher cost, exome and genome sequencing carry a higher risk of returning unwanted results such as variants of uncertain significance and secondary findings (disease variants in genes that are unrelated to the dystonia). Testing fewer genes reduces this risk. It is also occasionally possible for exome sequencing to give a false-negative result: for example, if the region containing the patient’s variant has insufficient data for an accurate result.[132] This is less of an issue with genome sequencing. If a gene panel is requested, one including detection of deletions/duplications is preferable.
Chromosomal microarray detects large CNVs, which are a different set of variants from the small/intragenic variants primarily found on NGS testing. Therefore, it could be appropriate to order microarray testing along with the other tests. If practical considerations limit the number of tests that can be done, it may be preferable to request one of the NGS-based tests, which have higher yields, and reserve microarray for later.
If gene panel testing is non-diagnostic, it is appropriate to move on to exome or genome sequencing. Exome sequencing has been shown to increase the diagnostic rate in patients who already had negative gene panels.[2]
If exome or genome sequencing is non-diagnostic (particularly with a variant of uncertain significance), it is reasonable to request re-analysis of the patient’s result approximately every 2 years.[133] Re-analysing the patient’s sequence in light of new information about disease genes and variants can sometimes allow their result to be revised to a more clinically useful classification such as disease-causing or benign.
If a genetic diagnosis is made, disease-specific treatment and counselling can be provided based on the known features of the disease.
If a complex dystonia syndrome is diagnosed, the patient can be screened for any systemic manifestations of the disease.
For conditions where treatment modifies the course of the disease, genetics consultation may be appropriate to determine whether any family members should undergo presymptomatic testing.
In some conditions, there are medical risks to heterozygotes that warrant evaluation of parents or additional family members. For instance, the mother of a patient with an X-linked condition could be at risk for later-onset symptoms. The same would apply for a parent carrying the same disease variant as the proband in an autosomal-dominant condition with variable expressivity.
Knowing the disease variant in the patient allows targeted testing for that specific variant in other family members. Parents can be offered genetic counselling to discuss their options for carrier testing, pre-implantation testing, and/or prenatal testing in future pregnancies.
Use of this content is subject to our disclaimer