Aetiology
Arginine vasopressin deficiency (AVP-D; previously known as central diabetes insipidus) arises from an absolute or relative deficiency of arginine vasopressin (AVP); AVP resistance (AVP-R; previously known as nephrogenic diabetes insipidus) results from a renal insensitivity or resistance to AVP. Both mechanisms reduce the permeability of the ducts within the nephron to water, reducing water resorption and thus increasing renal water loss. Causes of AVP-D or AVP-R can be genetic or acquired.[7]
AVP-D - acquired
Pituitary surgery: trans-sphenoidal pituitary surgery, usually for a pituitary adenoma, is a common cause of AVP-D.[3] Pituitary adenomas themselves do not present with AVP-D; the syndrome only manifests after surgical intervention. If AVP-D occurs in the presence of a sellar mass on imaging, an alternative diagnosis, such as craniopharyngioma, germinoma, pituitary metastases, or a granulomatous process should be considered.[3] AVP-D following pituitary surgery may be transient or permanent. It may rarely manifest as a triple-phase response: acute initial AVP-D, followed by a transient antidiuretic phase (presenting as hyponatraemia due to syndrome of inappropriate antidiuresis [SIAD]), subsequently progressing to permanent AVP-D.[12]
Craniopharyngioma: in contrast to most intracranial tumours, AVP-D is relatively common in patients with a craniopharyngioma - both preoperatively (8% to 35%) and postoperatively (70% to 90%).[13] Patients with craniopharyngiomas are also more likely to have associated primary and postoperative thirst abnormalities, which can exacerbate fluid balance and electrolyte problems.[14]
Post-traumatic head injury: around 20% of moderate to severe traumatic brain injuries are complicated by AVP-D.[3] While most cases resolve, permanent AVP-D has been reported in approximately 7% of cases.[15][16]
Pituitary stalk lesions: pituitary stalk involvement is common in Langerhans' cell histiocytosis (LCH); in one study, AVP-D was reported in up to 24% of patients with paediatric-onset LCH.[17] Other infiltrative conditions involving the pituitary stalk that can cause AVP-D include germinoma, intracranial metastases (particularly from breast or lung primaries), lymphoma/leukaemia, granulomatous disease (e.g., sarcoidosis and tuberculosis), lymphocytic infundibulohypophysitis, and IgG4-related disease.[1][3][18][19][20][21][22][23][24][25] Children and young adults with idiopathic AVP-D who have pituitary stalk thickening at presentation often develop a defined underlying diagnosis over time, as revealed through long-term follow-up.[26][27]
Autoimmune disorders: AVP-D is associated with other endocrine autoimmune disorders, including Hashimoto's thyroiditis, diabetes mellitus type 1, and systemic lupus erythematosus.[3][28] Autoantibodies to arginine vasopressin-secreting cells (AVPcAb) have been reported in 33% of patients with idiopathic (non-structural) AVP-D.[28]
Central nervous system infections: AVP-D may develop as a late complication of meningitis or encephalitis.[4][21][22][29][30]
Subarachnoid haemorrhage involving the anterior communicating artery (which supplies the anterior hypothalamus) can result in AVP-D and abnormalities in thirst.[31] AVP-D occurs in 15% of non-traumatic subarachnoid bleeds.[3]
Drugs: phenytoin is a possible cause of AVP-D, as is temozolomide (an oral chemotherapy drug used primarily to treat certain types of brain tumours).[32][33]
Coronavirus disease 2019 (COVID-19) has been linked to rare instances of new-onset AVP-D in a small number of case reports.[3][34][35] Proposed mechanisms include direct viral invasion of the hypothalamus or posterior pituitary via ACE2 receptors, immune-mediated inflammation damaging AVP-producing neurons, and vascular or hypoxic injury to the neurohypophyseal axis during severe illness.[36][37] In some cases the condition appears transient, suggesting reversible functional disruption.
Carbon monoxide poisoning is a rare cause of AVP-D.[38]
AVP-D - congenital/inherited
Familial AVP-D accounts for approximately 1% to 5% of all cases of AVP-D. It is usually an autosomal dominantly inherited condition due to a mutation in the AVP-NPII gene located on chromosome 20p135. This condition often has many family members presenting with clinical AVP-D early on in life.[39]
Congenital malformations involving the pituitary or hypothalamus and midline forebrain defects can result in AVP-D.[40] Examples include septo-optic dysplasia, agenesis of the corpus callosum, empty sella syndrome, and pituitary hypoplasia.[3]
AVP-D may occur as a component of Wolfram syndrome (WS) (also called DIDMOAD [diabetes insipidus, diabetes mellitus, optic atrophy, and deafness] syndrome), an autosomal-recessive, progressive neurodegenerative disorder characterised by diabetes mellitus and optic atrophy, with varying frequency of AVP-D and sensorineural deafness.[41] Genetic studies have identified two subtypes of WS.[42] WS1 is caused by loss-of-function mutations in the WFSI gene on chromosome 4p16. WFSI encodes an 890 amino-acid glycoprotein, wolframin, which regulates endoplasmic reticulum stress and calcium homeostasis. Loss of function of wolframin triggers apoptosis and cell death. Non-inactivating mutations of WFSI are associated with autosomal dominant sensorineural hearing loss. WSI may thus represent one extreme of a spectrum disorder. WS2 is also characterised by optic atrophy and diabetes mellitus. WS2 is caused by loss-of-function mutations in the CISD2 gene, which encodes a protein expressed in both the endoplasmic reticulum and outer mitochondrial membrane. Loss of function of CISD2 disrupts calcium flux, leading to autophagy and cell death.[41]
AVP-R - acquired
Drug-induced: lithium is a well-established cause of AVP-R, with studies reporting its occurrence in up to 40% of patients receiving long-term lithium therapy, and some estimates suggesting rates as high as 85%.[9][10] The mechanism involves lithium’s disruption of aquaporin-2 (AQP2) channels in the renal collecting ducts. AVP-R may be irreversible, persisting even after lithium therapy is discontinued.[43] Other drugs that can precipitate AVP-R include demeclocycline, cisplatin, colchicine, gentamicin, rifampicin, and sevoflurane.[5][44]
Systemic disease, electrolyte imbalance, and post-obstructive uropathy: chronic kidney disease, renal sarcoidosis, and renal amyloidosis are recognised causes of AVP-R.[5][20] AVP-R may also be caused by hypercalcaemia and hypokalaemia; in such cases, it may be reversible with correction of the underlying electrolyte imbalance.[5] Ureteric obstruction is another recognised risk factor for AVP-R.[5] Increased hydrostatic pressure is thought to suppress AQP2 channel expression in the renal collecting ducts. This down-regulation can persist for up to 30 days following relief of the obstruction, which may explain the diuresis often observed after the release of severe urinary tract obstruction.[10]
AVP-R - inherited
Approximately 90% of familial AVP-R cases are due to loss-of-function mutations in the AVPR2 gene, which encodes the vasopressin V2 receptor responsible for mediating the antidiuretic effects of AVP in the renal collecting ducts. These mutations result in X-linked recessive inheritance.[5][10][45]
AQP2 is the AVP-dependent water channel that renders the distal collecting duct permeable to water and allows AVP-mediated distal renal water resorption. Loss-of-function mutations in the AQP2 gene produce autosomal recessive familial AVP-R.[5][10][45] Loss-of-function mutations in the urea transporter-B gene also produce autosomal recessive AVP-R.[39]
Pregnancy and AVP-D
The osmotic thresholds for thirst and AVP release are altered in pregnancy.[46] There is also a fourfold increase in metabolic clearance of AVP due to placental production of vasopressinase/oxytocinase. This can unmask previously unrecognised AVP-D. In addition, pregnancy may aggravate the severity of pre-existing AVP-R or AVP-D.[1][6][47] Transient gestational AVP-D has been reported in patients with pre-eclampsia, HELLP (haemolysis, elevated liver enzymes, and low platelet count) syndrome, and acute metabolic dysfunction-associated steatotic liver disease (previously known as non-alcoholic fatty liver disease), where impaired hepatic degradation of vasopressinase is a contributing factor.
Pathophysiology
Plasma volume and osmolality are maintained within a narrow range by an integrated neuro-humoral system coordinating the balance between thirst-directed drinking (fluid intake) and renal water loss (fluid output).[48][49]
The key regulator of renal water loss is the 9-amino acid peptide hormone arginine vasopressin (AVP), also known as antidiuretic hormone (ADH).[48] AVP is synthesised in magnocellular neurons within the supraoptic (SON) and paraventricular (PVN) nuclei of the hypothalamus. AVP is produced from a large precursor peptide. The primary transcript undergoes significant post-translational processing as it is trafficked through the magnocellular neuron axons that terminate within the posterior pituitary, where the hormone is stored and released. Each molecule of AVP is co-secreted with two fragments of the original precursor: copeptin and neurophysin II (NPII).[50] Processing is outlined diagrammatically below.
[Figure caption and citation for the preceding image starts]: Vasopressin: gene structure and post-translational processingFrom the personal collection of Dr Stephen Ball [Citation ends].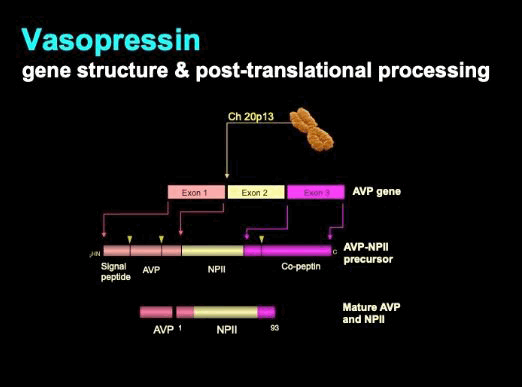
There is a linear relationship between plasma AVP concentration and plasma osmolality. Plasma osmolality is sensed by osmoreceptors within the hypothalamus, and these sensory afferents regulate AVP synthesis and release. AVP release is also regulated by circulating volume and blood pressure. This baroregulation can lead to AVP production and release at plasma osmolalities below the standard osmolar threshold of around 290 mmol/kg (290 mOsm/kg).[48][49]
[Figure caption and citation for the preceding image starts]: Physiology of AVP and thirstFrom the personal collection of Dr Stephen Ball [Citation ends].
Graded hyperosmolar stimulation defines very clearly the physiological characteristics of osmoregulated AVP production and thirst, represented in these two figures. The linear relationship between plasma osmolality and plasma AVP is further characterised by a functional osmolar threshold for AVP release; and a characteristic slope, which serves to define the sensitivity of the AVP response to osmolar stimulation. A very similar set of characteristics describe the relationship of thirst to plasma osmolality. Although the characteristics of osmoregulated AVP release and thirst vary between individuals (the range depicted here as the shaded areas or nomogram), the characteristics remain remarkably constant within an individual (depicted here as the solid line within the nomogram) over time.
AVP acts on type-2 AVP receptors (AVPR2) on the interstitial surface of renal distal collecting duct cells, increasing water permeability through the increased synthesis of aquaporin-2 water channels.[48] These are assembled and inserted into the apical membrane of collecting duct cells, resulting in AVP-dependent water resorption and concentration of urine.
AVP-D results from any condition that impairs the production, transportation, or release of AVP.
It is estimated that destruction of over 90% of the vasopressinergic neurons in the hypothalamus/posterior pituitary is necessary to deplete AVP sufficiently to cause hypotonic polyuria.[3]
AVP-R results from conditions that impair the renal collecting ducts' ability to respond to AVP.
Both AVP-D and AVP-R are characterised by impaired renal water re-absorption, resulting in the production of excessive, hypotonic (dilute) urine (polyuria), typically exceeding 3 litres (or >50 mL/kg) per day.[3] This is accompanied by significant thirst and increased drinking (polydipsia), as central osmo-sensing and peripheral baro-sensing drive central thirst and thirst-dependent behaviours to maintain circulating volume and osmolar status.
Patients with AVP-D or AVP-R due to a non-traumatic aetiology generally have an insidious onset of symptoms. Patients with AVP-D following traumatic brain injury or pituitary surgery typically experience a more rapid onset of symptoms. The severity of polyuria in AVP-D corresponds to the extent of neuronal damage, with complete destruction causing total AVP deficiency and marked polyuria, while partial damage with residual AVP secretion results in partial AVP-D and milder symptoms.[3]
Pregnancy is associated with a number of changes in salt and water regulation.[46] Transient AVP-D may develop as a consequence of a decreased osmotic threshold for thirst and AVP release, and a decrease in plasma osmolality. There is also a fourfold increase in metabolic clearance of AVP due to placental production of vasopressinase/oxytocinase in the second or third trimester.[3] In addition, pregnancy may aggravate the severity of pre-existing AVP-D or AVP-R.[1][6][47]
Classification
Polyuria-polydipsia syndromes
Primary polydipsia:
A disorder marked by an abnormally heightened sensation of thirst not caused by physiological triggers like dehydration or AVP dysfunction. It is primarily a condition of increased thirst sensation, often linked to psychological or psychiatric factors, and occasionally associated with structural abnormalities within the brain. Patients typically report excessive fluid intake followed by polyuria. This condition generally indicates a disturbance of thirst regulation rather than a defect in AVP secretion or function.
May be produced by drugs that cause a dry mouth, be associated with psychiatric syndromes, or be habitual.[1]
Psychogenic polydipsia is a subtype of primary polydipsia, characterised by excessive fluid intake driven by psychiatric conditions. It is most commonly seen in patients with schizophrenia, anxiety disorders, or other mental health disorders.
AVP-D:
Due to defective synthesis or release of arginine vasopressin (AVP) from the hypothalamo-pituitary axis.[1]
May be congenital (inherited) or acquired (e.g., neurosurgical procedures for tumours, trauma, autoimmune, infiltrative, vascular).[3][4]
AVP-R:
Due to renal insensitivity or resistance to AVP, with a resultant lack of permeability of the collecting duct to water.[1]
May be congenital (inherited) or acquired (e.g., drug-induced, particularly lithium; hypercalcaemia, hypokalaemia; obstructive uropathy).[5]
Transient AVP-D or AVP-R in pregnancy (previously known as gestational diabetes insipidus) can occur due to accelerated metabolism of AVP caused by placental production of cysteine aminopeptidases.[6]
Use of this content is subject to our disclaimer