Aetiology
Pancytopenia may be due to decreased bone marrow cell production or bone marrow failure, clonal disorders of haematopoiesis, increased non-immune-mediated destruction or sequestration, or immune-mediated destruction of blood cells. [Figure caption and citation for the preceding image starts]: Table of aetiologies for pancytopenia (SLE: systemic lupus erythematosus, CMV: cytomegalovirus, EBV: Epstein-Barr virus, GVHD: graft-versus-host disease)From the collection of Jeff K. Davies [Citation ends].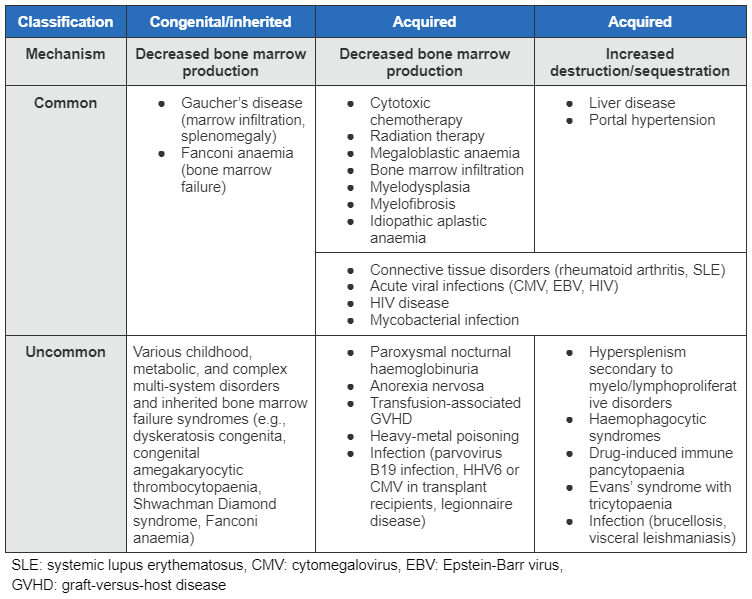
Decreased bone marrow production
The bone marrow is the site of production of red blood cells, white blood cells, and megakaryocytes, from which platelets arise. Once the cells are made, they are released into the peripheral circulation. This process requires adequate haematopoietic stem cell activity and a functional bone marrow stromal environment. The high proliferative rate of the marrow requires adequate nutritional status, particularly vitamin B12 and folic acid, and trace amounts of other elements.
Chemotherapy
The most common causes of transient pancytopenia in all age groups are cytotoxic chemotherapy and radiotherapy.
Chemotherapy-related pancytopenia rarely presents a diagnostic dilemma and usually resolves within 1 to 2 weeks (unless there is an unrecognised inherited bone marrow failure syndrome). Some individuals may have known or unknown proliferative defects or particular pharmacogenetics, which may predispose them to more severe and longer-duration pancytopenia. Some regimens are associated with significantly longer periods of pancytopenia. Longer-than-expected post-chemotherapy-related pancytopenia should be investigated.
Megaloblastic anaemia
Although most cases of megaloblastic anaemia cause macrocytic anaemia without leukopenia or thrombocytopenia, severe megaloblastic anaemia can result in pancytopenia. Megaloblastic anaemia most commonly arises from deficiency of vitamin B12 (e.g., pernicious anaemia, an autoimmune condition where autoantibodies interfere with the function of intrinsic factor, which is required for absorption of vitamin B12 within the gastrointestinal tract). Less commonly, B12 deficiency is caused by dietary deficiency (in vegans) or by malabsorption in the gut.
Folic acid deficiency, almost always dietary in origin, also results in megaloblastic anaemia.
Bone marrow infiltration
Infiltration of the bone marrow is a common cause of pancytopenia and commonly results from malignant disease. In general, the infiltrate is cellular and may be of haematological origin (e.g., acute myeloid and lymphoblastic leukaemia, myeloma, non-Hodgkin's lymphoma, hairy cell leukaemia, chronic lymphocytic leukaemia, and myelofibrosis) or non-haematological malignant origin (e.g., breast, lung, kidney, prostate, and thyroid).
In children, pancytopenia can be caused by neuroblastoma, rhabdomyosarcoma, Ewing's sarcoma, and retinoblastoma.[1]
Lysosomal storage disorders
Lysosomal storage disorders (e.g., Gaucher's disease) can infiltrate the marrow, resulting in pancytopenia. The infiltrate may be largely reticulin fibrosis, which is also associated with malignant conditions.
Patients with Gaucher's disease may have massive splenomegaly and functional hypersplenism in addition to infiltration of the bone marrow.[2]
Other causes
Rarer causes of pancytopenia arising from decreased bone marrow production of blood cells include anorexia nervosa, transfusion-associated graft-versus-host disease in immunosuppressed patients, and heavy metal poisoning (e.g., arsenic).[3]
Infections such as HIV have been associated with pancytopenia secondary to underproduction (frequently affecting only red cell production), as has parvovirus in individuals with specific predisposing conditions (haemolytic anaemia; most prominently sickle cell anaemia and hereditary spherocytosis).[4]
Clonal disorders of haematopoiesis
Myelodysplastic syndrome (MDS) is a common acquired clonal disorder of haematopoietic cells, characterised by ineffective and dysplastic haematopoiesis and a propensity for evolution to acute myeloid leukaemia. Adults are typically in their 70s at the time of diagnosis.[5][6] In children, the term refractory cytopenias of childhood (RCC) is often used. Children with MDS/RCC often have an underlying germline predisposition (IBMFS, RUNX1, ANKRD26, ETV6, GATA2, and SAMD9/SAMDL9) to MDS in approximately 30% to 40% of cases.[7]
The bone marrow may be either hypercellular or hypocellular. In both cases, there is commonly peripheral blood pancytopenia. In addition to decreased or inadequate production of blood cells within the marrow, there is sometimes an immune-mediated mechanism contributing to the peripheral blood pancytopenia in MDS.
Paroxysmal nocturnal haemoglobinuria (PNH) is a rare (incidence rate of 5.7 per 1,000,000 person-years) acquired clonal disorder of haematopoietic cells, caused by somatic mutation of the X-linked phosphatidylinositol glycan A gene and resulting in deficient expression of glycosylphosphatidylinositol-anchored proteins.[8][9] PNH is clinically characterised by intravascular haemolysis and thrombosis, and evolution of pancytopenia is common (probably arising from a combination of decreased bone marrow production secondary to acquired defects in haematopoietic stem cells and cell destruction). There is an overlap in clinical and laboratory features between patients with PNH and those with idiopathic aplastic anaemia (IAA) and even MDS.[Figure caption and citation for the preceding image starts]: Hypoplastic myelodysplastic syndrome with dysplastic normoblastsMorris Edelman, MD and Peihong Hsu, MD [Citation ends].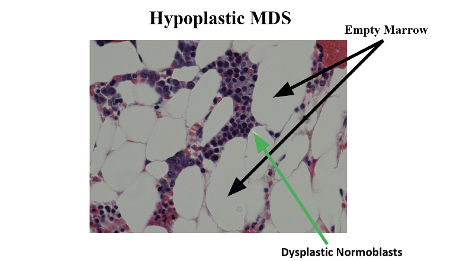
Bone marrow failure
Congenital and inherited bone marrow failure syndromes (IBMFS) most often present in childhood, although diagnosis in adulthood is increasing, secondary to awareness and greater testing.
Fanconi's anaemia: predominantly an autosomal recessive disorder (rare X-linked and dominant inheritance has been described) in which over 20 dysfunctional proteins result in decreased haematopoiesis and bone marrow failure.[10] Fanconi's anaemia is variably characterised by short stature, hyperpigmentation, skeletal anomalies, increased incidence of solid tumours and leukaemia, and an increased cellular sensitivity to DNA damaging agents.[11][12][13]
Dyskeratosis congenita (DC): inherited as X-linked, autosomal dominant or autosomal recessive disorder, arises from genetic lesions that compromise telomere integrity, with resulting loss of cell self-renewal and regeneration.[14] Mutations in 14 genes associated with telomere biology can be identified in the majority of patients with clinical features of classic DC.[15][16][17] Classic DC is defined by nail dystrophy, mucosal leukoplakia, and skin pigmentation changes, all ranging in severity from virtually non-existent to severe.[13] Other abnormalities include bone marrow failure, premature balding and grey hair, urethral strictures, excessive tear production, and pulmonary fibrosis.[18]
Idiopathic (acquired) aplastic anaemia (IAA): a rare acquired condition (2-6 cases per million general population). The diagnosis of IAA requires the presence of pancytopenia in combination with decreased bone marrow cellularity without infiltration or fibrosis.[19] IAA is therefore a diagnosis of exclusion and has to be differentiated carefully from hypocellular MDS, as well as congenital and IBMFS.[20] Some patients have an antecedent history of viral infection, hepatitis, or exposure to drugs. Severe IAA (where neutropenia and thrombocytopenia are more profound) is a life-threatening condition.
Other rare inherited single cell cytopenias: Diamond Blackfan anaemia (DBA), Shwachman Diamond syndrome (SDS), and amegakaryocytic thrombocytopenia (AMT) may evolve to pancytopenia.[13]
Using whole exome and whole genome sequencing, gene mutations seldom reported in inherited bone marrow failure have been identified in patients with bone marrow failure of suspected inherited origin but unresolved diagnosis.[21][Figure caption and citation for the preceding image starts]: Aplastic anaemia: normocellular bone marrow is shown on the left; and empty marrow, typical of congenital or acquired aplastic anaemia, is shown on the rightMorris Edelman, MD and Peihong Hsu, MD [Citation ends].
Increased destruction or sequestration
Most cases of pancytopenia that are accompanied by adequate bone marrow production of blood cells result from increased sequestration of blood cells within the spleen. Conditions that result in pancytopenia from functional hypersplenism include:
Liver disease (with associated portal hypertension) caused by alcoholic liver cirrhosis, chronic hepatitis B and C infection, autoimmune hepatitis, or idiopathic portal hypertension.
Myeloproliferative disorders (e.g., chronic myeloid leukaemia may present with massive splenomegaly resulting in pancytopenia despite adequate production of blood cells within the bone marrow). These conditions rarely occur in children.
Acute and chronic infections that result in hypersplenism (e.g., brucellosis and visceral leishmaniasis). Consideration of exposure and travel history is of particular relevance.
Haemophagocytic syndromes, a heterogeneous group of disorders characterised by increased macrophage or histiocyte activity within the bone marrow and other organs. Hepatomegaly and splenomegaly are common clinical features. Haemophagocytic syndromes may be categorised as primary (where the haemophagocytic syndrome dominates the clinical features of the condition, as in primary haemophagocytic lymphohistiocytosis [pHLH]), which are usually genetic in origin, or may be reactive to systemic conditions with a range of other clinical features (e.g., autoimmune disorders, T-cell lymphoma, often referred to as macrophage activation syndrome). These distinctions may be difficult to discern in some instances, but genetic testing is available for a number of the genes implicated in pHLH.[22]
Immune-mediated destruction of blood cells
Drug-induced immune pancytopenia arises when antibodies with cross-reactivity for drug and haematopoietic cells are generated. This is associated most frequently with quinine, sulfonamides, methotrexate, and rifampicin.[23]
Immune pancytopenia may be seen in up to 20% of patients with Evans syndrome (classically the combination of autoimmune thrombocytopenia and haemolytic anaemia), which is seen more commonly in children than in adults.[24] A significant number of people with Evans syndrome have underlying autoimmune lymphoproliferative syndrome.
Autoimmune lymphoproliferative syndrome (ALPS) is an inherited disorder resulting from mutations that inhibit apoptosis in the regulation of the immune response. Mild cases have been reported, suggesting that the incidence is significantly understated. ALPS is characterised by usually benign lymphoproliferation (lymphadenopathy and splenomegaly) and autoimmunity, most often directed towards cells of the myeloid lineage (erythrocytes, granulocytes, and platelets), although other targets are less commonly involved (e.g., autoimmune hepatitis).[25]
Combination pancytopenia
Many conditions associated with pancytopenia result from a combination of decreased bone marrow production and increased destruction or sequestration of blood cells. They include:
Connective tissue disorders (most commonly rheumatoid arthritis and systemic lupus erythematosus)
Acute cytomegalovirus infection
Mycobacterial infection
Infectious mononucleosis
HIV
Felty syndrome (rheumatoid arthritis, splenomegaly, and neutropenia).
Use of this content is subject to our disclaimer