Aetiology
Alport syndrome is one of several conditions caused by mutations in genes encoding type IV collagen. It is due to mutations in the COL4A3, COL4A4, and COL4A5 genes.[1] They encode the alpha-3, alpha-4, and alpha-5 chains of type IV collagen, which is a major component of the glomerular basement membrane. The glomerular basement membrane is unique because of its thickness (300 to 350 nanometre) and its position between two cell layers, podocytes, and endothelial cells. Other components of the glomerular basement membrane are laminin, nidogen, and heparin sulphate proteoglycans. Each type IV collagen molecule is composed of three sub-units or alpha chains that intertwine to form a helical structure. These collagen chains are characterised by a repeating Gly-X-Y sequence with a non-collagenous domain at the C-terminus. Six different genes, COL4A1 to COL4A6, encode the different alpha chain isoforms, alpha-1(IV) to alpha-6(IV). Alpha-1(IV) and alpha-2(IV) are ubiquitously expressed, while alpha-3(IV), alpha-4(IV), and alpha-5(IV) are restricted to the glomerular basement membrane, distal tubular basement membrane, Descemet membrane (thin hyaline membrane between the substantia propria and endothelial layer of the cornea) and Bruch membrane (inner layer of the choroid, separating it from the pigmentary layer of the retina), anterior lens capsule, lung, and cochlea. This accounts for the phenotypic features seen in Alport syndrome in the kidney, ear, and eye. Alpha-6(IV) is found in the epidermal basement membrane.
The COL4 genes are found as pairs in a 'head to head' orientation, COL4A1 to COL4A2 on chromosome 13, COL4A3 to COL4A4 on chromosome 2, and COL4A5 to COL4A6 on the X chromosome. Mutations in COL4A1 and COL4A2 are associated with brain small vessel disease manifesting as porencephaly (OMIM 120130), while mutations in COL4A1 also cause other diseases including hereditary angiopathy with nephropathy, aneurysms, and muscle cramps (HANAC syndrome).[19] Only mutations in COL4A3, COL4A4, and COL4A5 have been described in Alport syndrome, as well as a contiguous gene deletion of COL4A5 to COL4A6.[15] Mutations cause loss of expression or mis-assembly of the alpha chains and hence failure of assembly and maturation of a normal basement membrane collagen network.
Other phenotypes associated with variants in COL4A3, COL4A4, and COL4A5 include persistent dysmorphic haematuria, persistent proteinuria, steroid resistant nephrotic syndrome, focal segmental glomerulosclerosis, and end-stage kidney disease of unknown cause.[16]
[Figure caption and citation for the preceding image starts]: Classifications of Alport syndrome discussed in this topic with associated gene mutationsCreated by BMJ Knowledge Centre with information supplied by Richard N. Sandford [Citation ends].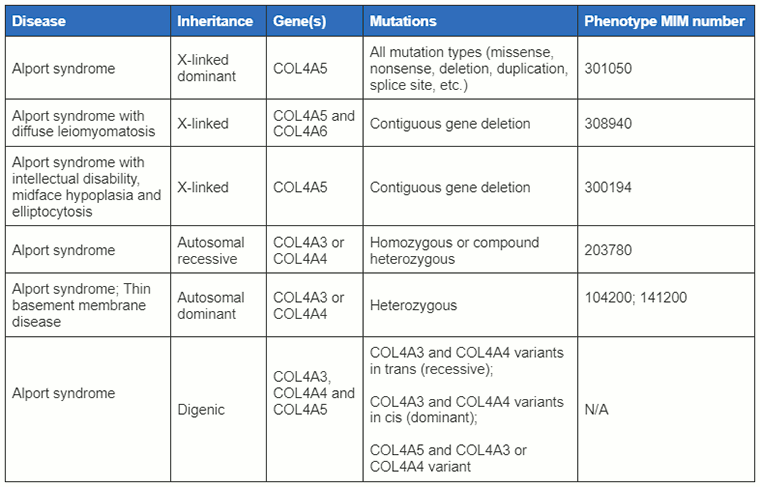
Pathophysiology
During early development alpha-1(IV) and alpha-2(IV) isoforms predominate in the glomerular basement membrane before switching to mainly alpha-3(IV), alpha-4(IV), and alpha-5(IV). These isoforms are thought to enhance the ability of the glomerular basement membrane to resist proteolytic degradation. Loss of these chains also leads to a compensatory increase in expression of other type IV, type V, and type VI collagens. Therefore, abnormalities in these components cause ultra-structural damage to the basement membrane, which manifests as thinning, thickening, splitting, and lamellation, the classic features seen under electron microscopy. This presumably results in the loss of the normal permselectivity of the glomerular basement membrane and the subsequent development of haematuria, proteinuria, glomerulosclerosis, and interstitial nephritis.
Classification
Genetic classification
Alport syndrome is a genetically heterogeneous disease caused by mutations in genes encoding different alpha chains of type IV collagen, namely COL4A3, COL4A4, and COL4A5.[1][5] It is one of a group of inherited disorders characterised by abnormalities in the glomerular basement membrane. Approximately 85% of cases are inherited as an X-linked trait and are due to mutations in the COL4A5 gene located at Xq22.3. The remaining 15% of cases are autosomal recessive and due to mutations in the COL4A3 and COL4A4 genes. Rare autosomal dominant and autosomal digenic cases are also due to mutations in the COL4A3 and COL4A4 genes.
X-linked Alport syndrome (XLAS)
XLAS is the most common type of Alport syndrome.[4] All male patients have haematuria and develop chronic kidney disease. There is considerable allelic heterogeneity. Genotype-phenotype correlations have been described with 90% of male patients having loss-of-function mutations (large deletions, non-sense and frame-shifting mutations) and reaching chronic kidney disease by 30 years of age.[4][6]
Haematuria develops in over 95% of females with XLAS with some developing chronic kidney disease in later life.[7] A family history may be absent in about 10% of cases suggesting a de novo mutation.
Autosomal recessive Alport syndrome (ARAS)
ARAS in a family is suggested by the presence of severe early disease in males and females, isolated haematuria in both parents and parental consanguinity.[8]
Autosomal dominant Alport syndrome (ADAS)
ADAS is rare and suggested by a relatively mild phenotype and slow progression to chronic kidney disease in affected patients.[9] The diagnostic term ADAS is not universally accepted for individuals heterozygous for a single COL4A3 or COL4A4 variant, as the majority will have haematuria only and a very low risk of developing chronic kidney disease, and as a result may not meet the criteria for Alport syndrome. The children of individuals with heterozygous ADAS also do not show a 50% likelihood of developing Alport syndrome, which is implied by the term ‘autosomal dominant’.[10]
Autosomal digenic Alport syndrome
This is rare and has a phenotype between ARAS and ADAS.[11]
Alport syndrome with diffuse leiomyomatosis (ASDL)
In ASDL, genital leiomyomas are often seen in females. There may also be a childhood history of cataracts, dysphagia, cough, altered bowel habit or recurrent bronchitis. However, there is often no family history.
Deletion in COL4A5 and COL4A6 is detected by multiplex ligation-dependent probe amplification and/or fluorescence in situ hybridisation (FISH) (if the deletion is large).
Alport syndrome with learning disability
This is classified as the presence of nephropathy with learning disability. There is often no family history but the characteristic facial appearance of mid-face hypoplasia is seen along with elliptocytosis on blood film.[12]
Whole gene COL4A5 micro-deletion is detected by FISH.
Use of this content is subject to our disclaimer