Approach
Sickle cell anaemia is usually diagnosed by neonatal screening.
Diagnosis is uncommon in childhood or adulthood, although it may occur. Most presentations to primary care physicians or hospitals are for complications. The most common complication is the vaso-occlusive crisis (also known as a painful crisis). The complication that frequently causes death in adults and children is acute chest syndrome.
Antenatal diagnosis
When both parents carry the recessive sickle cell gene, there is a 1 in 4 chance that their offspring will inherit two recessive alleles, causing sickle cell anaemia.
Counselling and antenatal diagnosis may be considered if both parents are either carriers or affected by sickle cell disease (or the woman is a carrier or affected and the status of the father is unknown). Available tests for patients who are pregnant include chorionic villus sampling (typically performed at 10-12 weeks' gestation in the US; 11-14 weeks' gestation in the UK) and amniocentesis (typically performed after 15 weeks' gestation).[20][21] These are invasive tests and carry a risk of fetal loss.
Non-invasive prenatal testing (NIPT) of cell-free fetal DNA in maternal circulation is emerging as a potential antenatal screening test for haemoglobinopathies such as sickle cell disease and thalassaemia.[16]
Diagnosis in neonates
The primary method by which neonates are diagnosed in the US is a screening programme. All states practise universal neonatal screening to ensure that all infants with sickle cell disease are identified.[22] Blood for screening is usually taken by heel-stick sample (within 48 hours of birth) and haemoglobin evaluation carried out (ideally within 24 hours of collection).[23][24]
Newborn screening is typically performed using haemoglobin isoelectric focusing (Hb IEF) or high-performance liquid chromatography (HPLC) fractionation. Confirmatory testing should be performed by an alternative method (IEF, HPLC, electrophoresis, or DNA sequencing); timely confirmation is important (no later than aged 3 months) to ensure prompt initiation of antibiotic prophylaxis.[15][25]
DNA testing is highly accurate and often used for confirming an abnormality found by haemoglobin studies. It may be used in place of haemoglobin studies in some screening programmes.[25][26][27][28]
UK newborn blood spot (NBS) screening
Worldwide approaches to neonatal screening vary. In the UK, all newborns are offered screening for sickle cell disease as part of the NHS newborn blood spot screening programme. A heel-stick sample is taken on day 5 after birth (or earlier if there is a family history of sickle cell disease).[29][15]
Diagnosis in infants
Infants do not usually manifest any signs and symptoms until they are 6 months old. However, very young infants with sickle cell disease not diagnosed by the screening programme may present with signs and symptoms of haemolysis (jaundice, pallor, or tachycardia) or splenic sequestration crisis (pallor, tachycardia, or shock).
From about age 4 months onwards, infants usually present with findings of:
Swelling of the joints, especially dactylitis[Figure caption and citation for the preceding image starts]: Hand-foot syndrome in patient aged 14 months with homozygous sickle cell diseaseFrom: Davies SC, Oni L. BMJ. 1997 Sep 13;315(7109):656-60 [Citation ends].
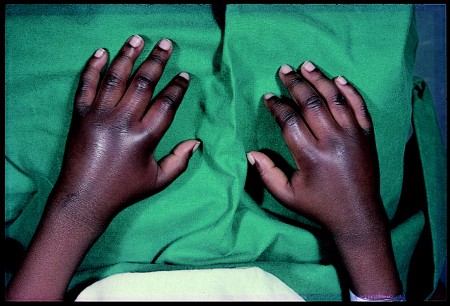
Leukocytosis in the absence of infection (the cause is not clear but is suggested to be due to splenic infarction)
Protuberant abdomen (due to an enlarged spleen), often with umbilical hernia
Cardiac systolic flow murmur secondary to anaemia
Maxillary hypertrophy with overbite due to extramedullary haematopoiesis, which occurs in some forms of the disease
Additionally, parents may report that their infant has been inconsolable for a long period of time, has been refusing bottles, and has needed fewer nappy changes during the previous 2 days.
The diagnosis is made using Hb IEF. Haemoglobin solubility testing is not recommended in infants aged under 6 months because the high proportion of fetal haemoglobin in relation to adult sickle cell haemoglobin in a newborn's blood may affect the results.
Diagnosis in children
If a child has not been diagnosed by the neonatal screening programme, the most common presenting complication is dactylitis, followed by recurrent episodes of pain and infection.
In older children, cellulose acetate electrophoresis at an alkaline pH is most commonly used to determine haemoglobin subtype. The diagnosis can be confirmed using an alternative method such as HPLC or a DNA-based assay.
Diagnosis in adults
It is unusual for a person with sickle cell disease to reach adulthood without being aware of his/her diagnosis. The diagnosis should be suspected in a patient who presents with unexplained haemolysis, with or without intermittent episodes of pain (vaso-occlusive crises). Patients may also present with avascular necrosis, retinal haemorrhage, or leg ulcers.
The first steps in diagnosis include review of the peripheral blood smear, followed by haemoglobin electrophoresis and confirmatory diagnosis with HPLC. In patients where a rapid diagnosis is required, the sickle solubility test can be done by adding a reducing agent (that decreases the oxygen content of the sample), which will cause sickle polymers to form in any cell with sickle haemoglobin. The test will detect any sickle haemoglobin and will be positive in patients with both sickle trait and sickle disease. Therefore, further confirmatory testing with HPLC or a DNA-based assay is needed.
Vaso-occlusive crisis
This common complication in children and adults causes severe pain and can be precipitated by cold, dehydration, infection, or ischaemia - for example, muscle ischaemia from strenuous exercise. The crisis may present as skeletal pain due to bone infarction or avascular necrosis, especially of the hip or shoulder.
The presentation of a skeletal vaso-occlusive crisis depends on the age of the patient, because the events are thought to originate in bone marrow. In children, red marrow is present in all bones, including small bones of the hand, which is consistent with the clinical findings of dactylitis. In older children, marrow is most commonly found in the epiphyses, and in adults it is limited to axial skeletal bones - for instance, spine, pelvis, skull, and the most proximal portions of the femur and humerus. This fits clinically with the observed incidence of infarcts in long bones increasing with age, especially in the femoral/humeral head.[Figure caption and citation for the preceding image starts]: Avascular necrosis of the femoral head in patient with heterozygous (haemoglobin SC) sickle cell anaemiaFrom: Davies SC, Oni L. BMJ. 1997 Sep 13;315(7109):656-60 [Citation ends].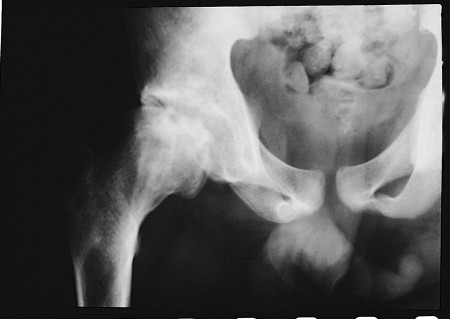
Other presentations may mimic an acute abdomen or pneumonia (acute chest syndrome).
Acute chest syndrome
Acute chest syndrome is a frequent cause of death in both children and adults. It can be clinically indistinguishable from pneumonia. The patient presents with chest pain, fever, dyspnoea, tachypnoea, hypoxaemia, and a new pulmonary infiltrate on chest x-ray.[Figure caption and citation for the preceding image starts]: Chest x-ray in acute chest syndromeFrom: Davies SC, Oni L. BMJ. 1997 Sep 13;315(7109):656-60 [Citation ends].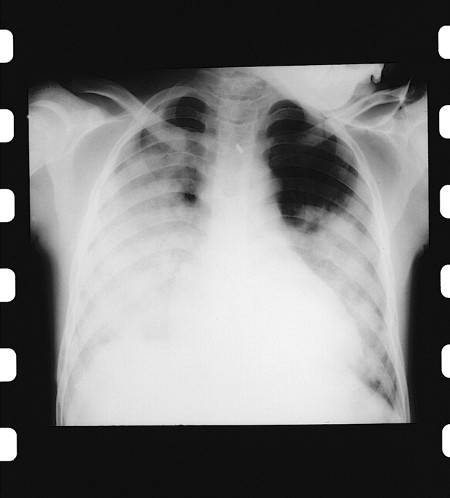
Other laboratory tests
Full blood count (FBC) with a peripheral blood smear is used in testing older children and adults to evaluate the number and quality of red blood cells, haemoglobin content, and white blood cell count. FBC is useful in establishing a baseline for on-going evaluation. It is widely available, inexpensive, and provides rapid results, but it is not diagnostic.
FBC should also be performed when a newborn screen is positive. It is important to remember that patients can present with vaso-occlusive pain and not have any change from baseline in their haemoglobin or reticulocyte count. [Figure caption and citation for the preceding image starts]: Red cells in sickle cell diseaseFrom the personal collection of Sophie Lanzkron, MD; used with permission [Citation ends].
Iron studies help distinguish haemolytic anaemia from iron deficiency anaemia in all patients and can also detect iron overload from multiple transfusions.
Bacterial cultures of blood, sputum, urine, stool, and/or pus should be obtained in patients with fever and in those who appear toxic.
Imaging
Plain x-rays are used to confirm the presence of bone infarction. However, radiological findings are localised to bones containing red marrow; therefore, the pattern of osseous changes is different in children and adults.
In children, red marrow is present in all bones including small bones of the hand, which is consistent with the clinical findings of dactylitis. In older children, red marrow is most commonly found in the epiphyses, and in adults it is limited to axial skeletal bones - for instance, spine, pelvis, skull, and the most proximal portions of the femur and humerus. This fits clinically with the observed incidence of infarcts in long bones increasing with age, especially in the femoral/humeral head.[Figure caption and citation for the preceding image starts]: Avascular necrosis of the femoral head in patient with heterozygous (haemoglobin SC) sickle cell anaemiaFrom: Davies SC, Oni L. BMJ. 1997 Sep 13;315(7109):656-60 [Citation ends].
Chest x-ray is performed if the patient has respiratory symptoms, fever, or chest pain.[Figure caption and citation for the preceding image starts]: Age distribution of clinical problems in sickle cell diseaseFrom: Davies SC, Oni L. BMJ. 1997 Sep 13;315(7109):656-60 [Citation ends].
How to take a venous blood sample from the antecubital fossa using a vacuum needle.
Use of this content is subject to our disclaimer