Etiologia
A causa da leucemia linfoide aguda (LLA) é desconhecida. Determinados fatores têm sido associados ao desenvolvimento da LLA.
Fatores genéticos
O diagnóstico da LLA em um gêmeo monozigótico (com <6 anos de idade) está associado a uma probabilidade de 10% a 15% de que o segundo gêmeo também desenvolva LLA.[12]
A LLA está associada à trissomia do cromossomo 21 e a outras doenças genéticas (por exemplo, síndrome de Li-Fraumeni, neurofibromatose, síndrome de Klinefelter, anemia de Fanconi, síndrome de Shwachman-Diamond, síndrome de Bloom e ataxia-telangiectasia).[13][14][15][16]
Há cada vez mais evidências que sugerem uma predisposição de linha germinativa à LLA.[14][17][18][19][20] As mutações das linhas germinativas ligadas à LLA são relatadas em aproximadamente 4% das crianças com LLA.[14]
Outros fatores
Fisiopatologia
Na LLA, a anormalidade genética de uma célula progenitora linfoide resulta em proliferação descontrolada e expansão clonal. Os linfoblastos leucêmicos se infiltram na medula óssea e em outros órgãos, o que prejudica seu funcionamento normal. Linfoblastos leucêmicos também podem circular no sangue periférico.
Os linfoblastos leucêmicos representam uma expansão clonal de uma única célula progenitora linfoide.[3][32][33][34] Os linfoblastos leucêmicos duplicam a maioria das características da célula progenitora linfoide.
As anormalidades genéticas na LLA incluem rearranjos cromossômicos (por exemplo, translocações), aneuploidia (número anormal de cromossomos) e mutações genéticas. Translocações cromossômicas ou aneuploidia são encontradas em 75% dos casos. As translocações são, com frequência, recorrentes e raramente classificadas como translocações aleatórias.[33][34][35][36]
LLA-B positiva para cromossomo Filadélfia (LLA-B Ph+)
Uma das translocações cromossômicas mais comuns e clinicamente importantes na LLA-B em adultos é a t(9;22)(q34;q11), que resulta no gene de fusão BCR::ABL1 no cromossomo 22 (ou seja, o cromossomo Filadélfia).[37] O gene de fusão BCR::ABL1 codifica uma tirosina quinase ativa que transforma células-tronco hematopoiéticas normais em células leucêmicas.[Figure caption and citation for the preceding image starts]: Translocação BCR::ABL1Do acervo do Dr. Han Myint e do Dr. Robert Chen; usado com permissão [Citation ends].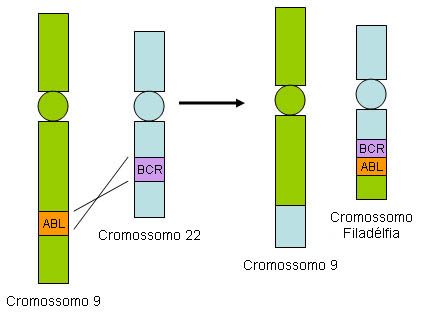
LLA-B tipo Filadélfia (LLA-B tipo Ph)
A LLA-B tipo Ph é um subtipo de alto risco que tem um perfil de expressão gênica similar à LLA-B Ph+, mas não tem o gene de fusão BCR::ABL1 (cromossomo Filadélfia).[37] A LLA-B tipo Ph compreende 10% e 13% dos casos de LLA-B de risco padrão e de alto risco na infância, respectivamente.[33]
A frequência da LLA-B tipo Ph aumenta com a idade, sendo ela responsável por >25% dos casos em adultos jovens.[37] A presença de LLA-B tipo Ph está associada a um desfecho desfavorável.[38][39]
Os pacientes com LLA-B tipo Ph podem ser classificados da seguinte maneira:[40]
Tipo I, fusões da classe ABL (ABL1, ABL2, CSF1R, PDGFRB)
Tipo II, receptor de eritropoetina (EPOR) ou rearranjos de JAK2
Tipo III, rearranjos de fator 2 semelhante ao receptor de citocina 2 (CRLF2) (geralmente acompanhados por mutações JAK2 e ativação do sinal JAK-STAT)
Tipo IV, outras mutações ativadoras da sinalização JAK-STAT (IL7R, FLT3, SH2B3, TYK2, IL2RB)
Tipo V, mutações incomuns da quinase diversas (NTRK3, DGKH)
Tipo VI, mutações da via RAS (KRAS, NRAS, PTPN11, NF1)
Tipo VII, nenhuma mutação nos genes da quinase.
Outras anormalidades genéticas
FLT3 e NOTCH1 foram identificados como genes com mutação na leucemia de linhagem mista (MLL)/hiperdiploide e na LLA-T, respectivamente.[41] Mutações CREBBP foram observadas em 18% das LLA recidivantes e podem oferecer resistência à terapia.[42] O gene PAX5 sofreu mutação em até 30% dos pacientes pediátricos com LLA.[43][44] As mutações de PHF6 são observadas em 38% das amostras de LLA-T em adultos.[45] As mutações de CDKN2A são observadas em 42% de todos os casos de LLA-T.[46]
Alguns rearranjos gênicos podem resultar em perda ou ganho de mutações de funções que envolvem os fatores de transcrição que participam do desenvolvimento hematopoiético. Um exemplo desse rearranjo gênico é a translocação cromossômica t(12;21)(p13;q22) que resulta no gene de fusão ETV6::RUNX1 (também conhecido como TEL::AML1).[1][2]
A perda ou inativação de genes supressores de tumores por meio de deleções e rearranjos gênicos (por exemplo, IKZF1, p16INK4) está associada com o desenvolvimento de LLA.[47][48] As mutações em IKZF1 podem ser preditoras de recidiva.[49] A deleção de IKZF1 com deleções concomitantes em CDKN2A, CDKN2B, PAX5 ou PAR1 (na ausência de deleção de ERG) define um subgrupo conhecido como "IKZF1 plus", que está associado a um prognóstico particularmente desfavorável.[50]
Algumas anormalidades genéticas recorrentes (por exemplo, BCR::ABL1, rearranjo de KMT2A, ETV6::RUNX1) foram incorporadas em sistemas de classificação de doenças da Organização Mundial da Saúde (OMS) e da International Consensus Classification (ICC) para subclassificar a LLA.[4][5] Consulte Classificação.
Classificação
5a edição da classificação de tumores hematolinfoides da Organização Mundial da Saúde: neoplasias linfoides.[4]
A classificação da LLA (e entidades) se baseia na linhagem (LLA-B ou LLA-T) e na presença de anormalidades citogenéticas/moleculares.
Leucemia linfoide B/linfoma:
Leucemia linfoide B/linfoma, sem outra especificação
Leucemia linfoide B/linfoma com alta hiperdiploidia
Leucemia linfoide B/linfoma com hipodiploidia
Leucemia linfoide B/linfoma com iAMP21
Leucemia linfoide B/linfoma com fusão BCR::ABL1
Leucemia linfoide B/linfoma com características semelhantes a BCR::ABL1
Leucemia linfoide B/linfoma com rearranjo de KMT2A
Leucemia linfoide B/linfoma com fusão ETV6::RUNX1
Leucemia linfoide B/linfoma com características semelhantes a ETV6::RUNX1
Leucemia linfoide B/linfoma com fusão TCF3::PBX1
Leucemia linfoide B/linfoma com fusão IGH::IL3
Leucemia linfoide B/linfoma com fusão TCF3::HLF
Leucemia linfoide B/linfoma com outras anormalidades genéticas definidas (por exemplo, rearranjos de DUX4, MEF2D ou ZNF384)
Linfoma/leucemia linfoide T:
Linfoma/leucemia linfoide T, sem outra especificação
Linfoma/leucemia linfoide de células T precursoras inicial
Classificação de Consenso Internacional (ICC) de neoplasias mieloides e leucemias agudas[5]
A classificação da LLA (e entidades) se baseia na linhagem (LLA-B ou LLA-T) e na presença de anormalidades citogenéticas/moleculares.
Linfoma/leucemia linfoide B aguda
LLA-B com anormalidades genéticas recorrentes
LLA-B com t(9;22)(q34.1;q11.2)/BCR::ABL1
com envolvimento apenas linfoide
com envolvimento multilinhagem
LLA-B com rearranjo de t(v;11q23.3)/KMT2A
LLA-B com t(12;21)(p13.2;q22.1)/ETV6::RUNX1
LLA-B, hiperdiploide
LLA-B, baixa hipodiploidia
LLA-B, quase haploide
LLA-B com t(5;14)(q31.1;q32.3)/IL3::IGH
LLA-B com t(1;19)(q23.3;p13.3)/TCF3::PBX1
LLA-B, semelhante a BCR::ABL1, rearranjo de classe ABL-1
LLA-B, semelhante a BCR::ABL1, JAK-STAT ativado
LLA-B, semelhante a BCR::ABL1, sem outra especificação
LLA-B com iAMP21
LLA-B com rearranjo de MYC
LLA-B com rearranjo de DUX4
LLA-B com rearranjo de MEF2D
LLA-B com rearranjo de ZNF384(362)
LLA-B com rearranjo de NUTM1
LLA-B com rearranjo de HLF
LLA-B com UBTF::ATXN7L3/PAN3,CDX2 ('CDX2/UBTF')
LLA-B com mutação IKZF1 N159Y
LLA-B com mutação PAX5 P80R
LLA-B, sem outra especificação
Entidade provisória: LLA-B, semelhante a ETV6::RUNX1
Entidade provisória: LLA-B, com alteração em PAX5
Entidade provisória: LLA-B, com mutação ZEB2 (p.H1038R)/IGH::CEBPE
Entidade provisória: LLA-B, com rearranjo semelhante a ZNF384
Entidade provisória: LLA-B, com rearranjo semelhante a KMT2A
Linfoma/leucemia linfoide T aguda
LLA inicial de células T precursoras com rearranjo de BCL11B
LLA inicial de células T precursoras, sem outra especificação
LLA-T, sem outra especificação
Entidade provisória: LLA-T, com desregulação de HOXA
Entidade provisória: LLA-T, com rearranjo de SPI1
Entidade provisória: LLA-T, com rearranjo de TLX1
Entidade provisória: LLA-T, com rearranjo de TLX3
Entidade provisória: LLA-T, com rearranjo de NKX2
Entidade provisória: LLA-T, com rearranjo de TAL1-2
Entidade provisória: LLA-T, com rearranjo de LMO1-2
Entidade provisória: LLA-T, BHLH, outro
O uso deste conteúdo está sujeito ao nosso aviso legal