Details
Complement deficiencies can be inherited or acquired, and may increase the risk of invasive bacterial infections or be associated with autoimmune disease. Acquired complement deficiencies may occur following infection (e.g., recurrent meningococcal or disseminated gonococcal infection) or in conjunction with chronic rheumatological or autoimmune disease (e.g., systemic lupus erythematosus or cryoglobulinaemia).
The complement system itself is an essential part of the innate immune system, with complement proteins moderating antibody actions, aiding in the processing and removal of immune complexes, and modifying T-cell and B-cell responses.
Diagnosis is based on clinical and/or family history and characteristic serological and molecular findings; only a few specialised laboratories provide comprehensive diagnostics.
Suspicious presentations include meningococcal meningitis in individuals aged over 5 years, recurrent bacterial infections, angio-oedema without urticaria, inflammatory disorders of the renal and ophthalmic system, and autoimmune manifestations.
The complement system plays a key role in host microbial defence and tissue homeostasis.[1][2] It complements the actions of specific antibodies and aids the removal of immune complexes and apoptotic cell debris.[3][4] It also modifies T-cell and B-cell responses by employing specific receptors on various immune cells to modulate adaptive immune response.[4] The complement system participates in haematopoiesis, lipid metabolism, reproduction, and tissue regeneration.[3] Multiple interactions connect complement to the clotting, fibrinolysis, and kinin systems.[5][6] Due to its powerful inflammatory potential, multiple regulatory proteins are necessary to prevent potential tissue damage.[7]
When overactive, the complement system can cause several inflammatory and life-threatening conditions, including sepsis, systemic inflammatory response syndrome, acute respiratory distress syndrome, and multiple organ failure after severe trauma, burns, or infections.[8][9] It is implicated in severe nephropathies as well as in neurodegenerative disorders, such as Alzheimer's disease, Guillain-Barré syndrome, and multiple sclerosis.[10][11][12][13] An over-activated complement system has also been recognised as a significant effector mechanism of reperfusion injury. Organ dysfunction may be caused by the inflammatory response induced by artificial surfaces in haemodialysis and extracorporeal circuits.[14] In these cases, this may lead to transient neutropenia, pulmonary vascular leukostasis, and more rarely, anaphylactic shock.
Disease states occur with deficiencies in complement proteins or defects of factors controlling, focusing, and limiting complement activation.[15][16][17][18][19]
The complement system comprises more than 50 complement proteins, including individual components of the cascade system, regulatory factors (many of which are cell-surface restricted), and receptors. They are present in plasma and other body fluids, and may also exert essential intracellular immune modulatory functions.[20][21]
Complement genes are distributed across different chromosomes.[22]
Most complement proteins are secreted by the liver and form part of the acute phase response. Other tissues (e.g., kidney, brain) are also able to produce complement proteins, such as fatty cells for factor D (adipsin). Certain complement components are proteases, which, on activation, cleave the next complement protein in the cascade sequence. The sequence of amplification steps shows similarities to that of the blood coagulation cascade.
The complement cascade can be activated by three main routes: the classical pathway (CP), the alternative pathway (AP), and the lectin pathway (LP).[23][Figure caption and citation for the preceding image starts]: Complement cascade: activation and regulation. Complement is activated via three pathways, the classical, the alternative, and the lectin pathways. This results in: (1) the generation of various pro-inflammatory peptides, which bind to specific receptors on multiple immune cells, as well as (2) the assembly of the membrane attack complex, C5b-9 (MAC). On each level of the cascade reaction, complement is tightly regulated by soluble and membrane-associated inhibitors.Adapted from a figure provided by Michael Kirschfink DVM, PhD; used with permission [Citation ends].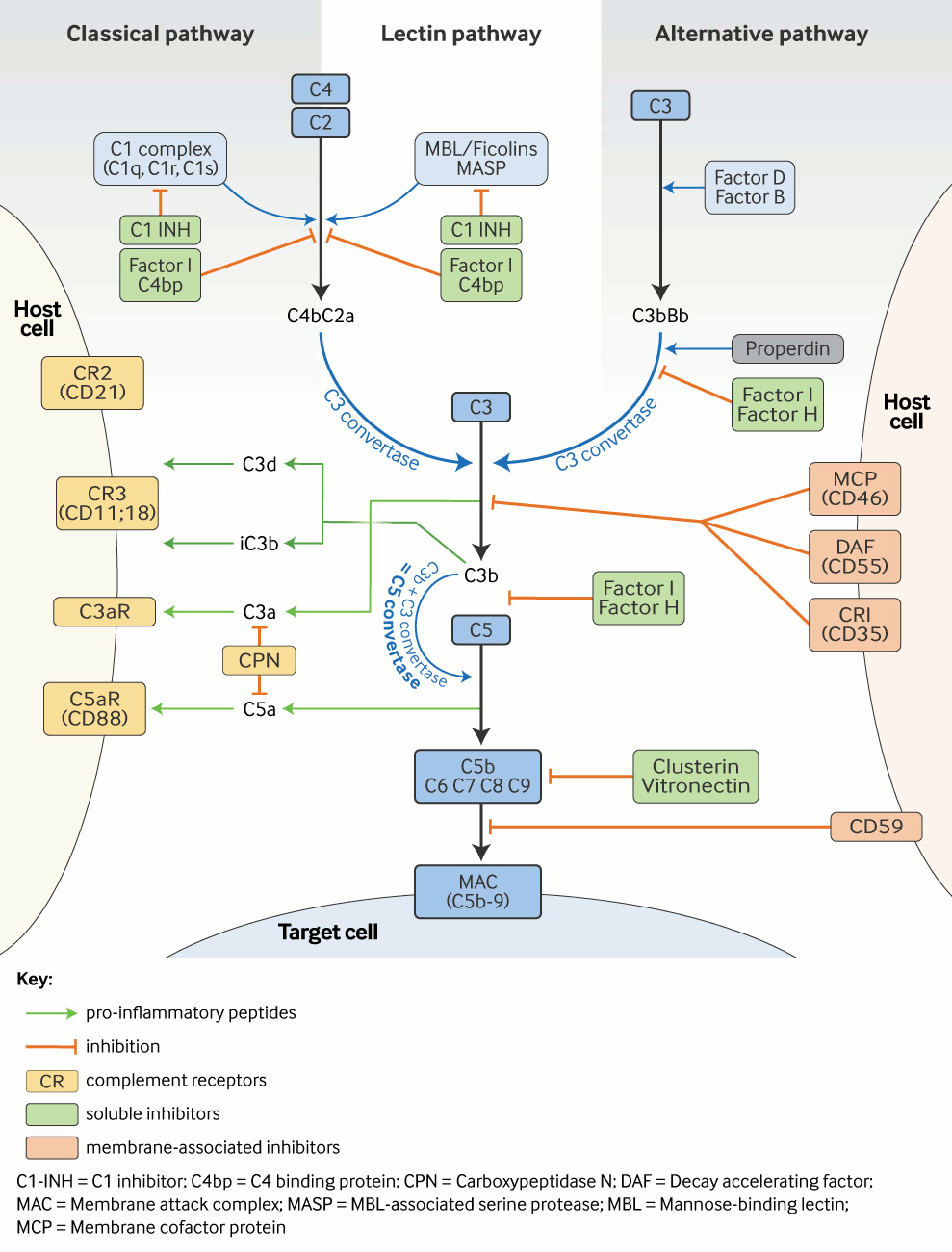
The CP serves as a key effector function of specific antibody responses, whereas the AP and LP, as part of the innate immune system, are important in first-line antibody-independent defence against microbial infections. The terminology of the complement system components relates to the sequence in which they were discovered, which explains why the cascade is not arranged in a logical numerical order. Component proteins and regulators of the AP are called factors (e.g., factor B, factor H).[24]
CP activation is primarily initiated by antibody (immunoglobulin)-derived mechanisms. The fragment crystallisable (Fc) regions of IgM and IgG antibodies (but not IgA) become structurally altered (e.g., when bound to a specific antigen within an immune complex or within a cryoglobulin). The CP is activated when C1 binds to the Fc region via its C1q moiety.
In the absence of antibodies, target-bound C-reactive protein (CRP) can also bind to C1q and activate the CP. The LP is initiated by the binding of ficolins or mannose-binding lectin (MBL, a well known opsonin and an acute phase reactant with structural similarities to C1q) to carbohydrate residues on pathogens and altered tissues. Similar to the C1 complex, MBL-carbohydrate binding leads to the activation of MBL-associated serine proteases (MASPs) that, like C1s, are able to cleave C4 and C2, thereby connecting the LP to the CP. By contrast with the CP, the AP is activated mainly by non-antibody (non-immunoglobulin) mechanisms. Permanent low-grade hydrolysis of C3 (C3[H₂O]) leads - upon binding of factor B and subsequent cleavage by factor D - to the generation of a fluid phase C3-convertase (C3b[H₂O]Bb), which is stabilised by properdin. In healthy states this activity is self-limited; however, if newly-cleaved C3 binds to pathogens or altered tissue, the AP response is amplified. The regulatory potential of the targeted cells determines whether a C3 convertase is formed on the surface, opsonisation occurs, and the cascade reaction is continued.
Once complement is activated by any of these pathways, enzyme complexes (C3 convertases) are generated that cleave C3 into 2 fragments (C3a and C3b). C3a is the smaller fragment. Like C5a, which is generated downstream the cascade, C3a is a pro-inflammatory signalling molecule (anaphylatoxin). Anaphylatoxins are chemoattractants. They recruit and activate multiple inflammatory cells, including neutrophils and mast cells. Receptors for C3b and its metabolic product iC3b on phagocytic cells allow removal of the opsonised targets. Potentially pathological immune complexes (containing antibody complexed with viral, bacterial, or autoantigens) activate the CP and C3b, which flags them for removal from the circulation by C3b-receptor-carrying erythrocytes and disposal by phagocytic cells in the reticulo-endothelial system. Upon C3b binding to the C3 convertase, a C5 convertase is generated that cleaves C5 into C5a and C5b. C5b together with C6, C7, C8, and C9 generate the lipophilic membrane attack complex (MAC), C5b-9, causing target cell death by cell membrane lysis.
Multiple regulatory proteins are necessary to ensure that potential complement-mediated tissue damage is prevented or at least limited.[25] Factor H and factor I regulate the AP, whereas C1-inhibitor (C1-INH) and C4 binding protein (C4bp) control the CP and LP. C3 convertases are inherently unstable, with short half-lives, which helps limit and control complement activation. Membrane cofactor protein (MCP, CD46), and decay accelerating factor, (DAF, CD55), control C3 activation on the cell surface. Excess MAC-mediated complement lysis is prevented by the soluble inhibitors clusterin and vitronectin, and the membrane-associated MAC-inhibitory protein (CD59).[Figure caption and citation for the preceding image starts]: Complement cascade: activation and regulation. Complement is activated via three pathways, the classical, the alternative, and the lectin pathways. This results in: (1) the generation of various pro-inflammatory peptides, which bind to specific receptors on multiple immune cells, as well as (2) the assembly of the membrane attack complex, C5b-9 (MAC). On each level of the cascade reaction, complement is tightly regulated by soluble and membrane-associated inhibitors.Adapted from a figure provided by Michael Kirschfink DVM, PhD; used with permission [Citation ends].
Complete defects have been described for all complement proteins except serum carboxypeptidase N. Secondary deficiencies result from complement consumption as a result of inflammation, autoantibodies (e.g., C3/C5 nephritic factors; and autoantibodies against C1q, C1-inhibitor, or factor H), decreased synthesis, and/or increased catabolism or protein loss syndromes. Gain-of-function variants of certain complement components (e.g., C3, C2) have been identified and associated with complement over-activation in certain forms of nephropathies.[26][27][28]
Complement deficiency states and associated features
Components
C1q: systemic lupus erythematosus (SLE)-like (occurs in >90% of patients with C1q deficiency), infections
C1r/s (mostly combined): SLE-like, rheumatoid arthritis (RA), infections
C4 (C4A, C4B): SLE-like, infections (homozygous: usually clinically apparent; heterozygous: often clinically inapparent)
C2: SLE-like, RA, infections (pneumonia), vasculitis; often clinically inapparent
C3: pyogenic infections
C5: meningitis (Neisseria), SLE
C6: meningitis (Neisseria), SLE
C7: meningitis (Neisseria), SLE
C8 alpha-gamma/C8 beta: meningitis (Neisseria), SLE
C9: neisserial infections (mostly asymptomatic)
Factor B: neisserial infections
Factor D: neisserial infections
Mannose-binding lectin (MBL): bacterial infections (mostly asymptomatic)
Ficolin 3 (H-ficolin): respiratory infections, necrotising enterocolitis
MBL-associated serine protease 2 (MASP-2): respiratory infections
Regulators
C1-inhibitor: hereditary angio-oedema
Properdin: meningitis (Neisseria)
Factor H: infections, atypical haemolytic uraemic syndrome (aHUS)/C3 glomerulopathy (C3G), aHUS, C3G/membranoproliferative glomerulonephritis (MPGN)
FHR1 (FHR3): aHUS, RA, SLE (often associated with anti-factor H autoantibodies and deficiency of CFHR [complement factor H-related] proteins, causing susceptibility to autoantibody-mediated aHUS)
Factor I: infections (sepsis, meningitis, pneumonia), aHUS
CD46/MCP (membrane cofactor protein): aHUS
CD55/DAF (decay accelerating factor): severe enteropathy (protein-losing enteropathy), paroxysmal nocturnal haemoglobinuria (somatic mutation of PIGA gene)
CD59: Guillain-Barré syndrome-like symptoms, haemolysis, paroxysmal nocturnal haemoglobinuria (somatic mutation of PIGA gene)
Receptors
CR3 (CD18/CD11b): leukocyte adhesion deficiency
CR4 (CD18/CD11c, LFA-1): leukocyte adhesion deficiency
Complement deficiencies represent approximately 3% to 5% of all primary immunodeficiencies, but may be as high as 10%.[29][30][31] European Society for Immunodeficiencies (ESID) reporting website and interactive reporting tool Opens in new window
Inherited complement deficiency has been calculated to have a prevalence of about 0.03%, excluding mannose-binding lectin (MBL) deficiency, which is estimated to occur in about 5% of the general white population.[29] However, as with all inborn errors of immunity/primary immunodeficiencies, a significant number of patients with complement deficiency probably remain undiagnosed due to limited clinical and laboratory experience of the conditions. The most frequent complement deficiencies affect C2 and MBL, which most often remain clinically silent.
However, complement protein deficiencies are significantly more common in people with specific diseases. In systemic lupus erythematosus, 30% of patients have a pre-existing complement deficiency and in individuals with disseminated Neisseria infections it is thought to be around 20%.[32][33]
Presenting infections due to complement deficiency can include:
Recurrent pyogenic infections (e.g., deep abscess, osteomyelitis, pneumonia)
Bacteraemia
Recurrent meningococcal infection
Disseminated gonococcal infection
Neisseria bacteria (meningococcal and gonococcal) are particularly sensitive to complement-mediated attack. With the exception of recurrent Neisseria infections, patients with recurrent unexplained pyogenic bacterial infections should also be checked for other immune deficiencies including immunoglobulin or phagocyte deficiency, which are more prevalent than complement deficiency.
Some specific clinical presentations (warning signs) raise the possibility of a complement deficiency.[29] These include:
Meningococcal meningitis in individuals over the age of 5 years
Other recurrent bacterial infections, especially Pneumococcus
Angio-oedema without urticaria
Autoimmune manifestations
Inflammatory disorders of the renal and ophthalmic systems
Extragenital gonococcal infections
Inheritance is usually autosomal recessive, with some exceptions, such as properdin deficiency (which is X-linked), and C1-inhibitor (autosomal dominant).[15][34] Heterozygous carriers usually remain clinically silent, except for patients with age-related macular degeneration and certain nephropathies (atypical haemolytic uraemic syndrome, C3 glomerulopathy). Inherited complement deficiencies can be identified by taking an accurate medical history and performing extended laboratory analysis of the entire family.
C1-inhibitor deficiency and angio-oedema
The incidence of hereditary angio-oedema (HAE; also known as Quincke's oedema) with C1-inhibitor deficiency (HAE-C1-INH) is estimated as approximately 1:50,000.[35][36]
HAE manifests as acute, paroxysmal swelling of the lips, eyelids, and gastrointestinal tract (colic), and can be particularly dangerous if swelling is localised in the larynx and pharynx. Intestinal obstruction, urinary retention, and headache can be caused by oedema of other organs. Stress, trauma-related infections, and variations in hormones due to pregnancy, contraception, menstruation, or hormone replacement therapy can trigger angio-oedema. Attacks last up to 1 week and if not treated can lead to asphyxia in 25% to 40% of cases.[36]
There are two autosomal-dominant forms of HAE associated with a C1-inhibitor deficiency (HAE-C1INH). Inadequate C1-inhibitor production (HAE-1) is recognised in 80% of cases and the synthesis of dysfunctional C1-inhibitor (HAE-2) accounts for the remaining 20%.[37] Both forms typically show reduced C4 as a secondary consequence of classical pathway dysregulation. C1-inhibitor functional tests are abnormal in both types, while plasma levels of C1-inhibitor are decreased in HAE-1, but are normal or elevated in HAE-2.[35][38]
Bradykinin is the key mediator for vascular leakage contributing to angio-oedema. The much rarer acquired angio-oedema develops following the excessive catabolism of C1-inhibitor or blocking of autoantibodies.[39][40] It shows similar symptoms to those of HAE-1 and 2.[35]
Many patients present with clinical manifestations that are similar to HAE but present normal C1-inhibitor and C4 levels (HAE-nlC1INH). In these patients, gain-of-function mutations in the FXII gene (HAE-FXII), and deficiencies of plasminogen (HAE-PLG), kininogen (HAE-KNG1), myoferlin (HA-MYOF), and heparan sulfate-glucosamine (3-O-sulfotransferase 6) (HAE-HS3OST6) have been observed.[41]
C1-inhibitor and C1q may also be consumed as part of massive immune complex type diseases, such as urticarial vasculitis.[42]
See Urticaria and angio-oedema.
Deficiency of classical pathway components and SLE
A pre-existing complement deficiency or malfunction of classical pathway (CP) components including C1q, C4, C2, C3, or complement receptors (including C3b receptor) can predispose to SLE. The most prevalent and severe SLE is associated with C1, C2, or C4 deficiency.[43][44] More than three-quarters of individuals deficient in one of these proteins have SLE - more than 90% in cases of C1q deficiency.[43] Approximately 10% of individuals with C2 deficiency have SLE.[43] Autoantibodies against C1q (prevalent in a high percentage of patients with SLE) may neutralise C1q function and are considered of prognostic value, in particular for the development and activity of lupus nephritis.[45] Reduced complement-mediated elimination of immune complexes and apoptotic cells is characteristic of this disease and is most pronounced in patients with C1q genetic deficiency.[46][47] Monitoring CP haemolytic function and biochemical levels of C3 and C4, activation products (such as C4d, and C3d), and the anti-C1q autoantibody is of use (especially when SLE causes glomerulonephritis, clinical improvement is often accompanied by the restoration of normal C3 and C4 levels).[45][48]
Complement deficiencies in disseminated Neisseria infections
Deficiencies in C3, terminal components of the complement pathway (C5-C9), and the alternative pathway (AP) protein properdin are strongly associated with an increased incidence of invasive meningococcal infections, especially with rare serotypes (W, X, Y, Z).[49] This is indicative of the importance of cytolytic complement activity in host defence against Neisseriae. To identify such deficiencies, it is mandatory to start analysis by a global screening of the functional activity of both the classical and alternative pathways using either haemolytic assays (CH50, AH50) or a functional enzyme-linked immunosorbent assay (ELISA), which also includes the lectin pathway. Normal AH50 and CH50 levels with recurrent meningococcal infection may point to a properdin defect.
It is strongly recommended that patients are immunised against Neisseria meningitidis with a tetravalent conjugate vaccine.[15][50][51][52] It should be noted that not all disease-related serotypes are covered by such vaccines.
Complement dysregulation in nephropathies
Since the kidney is particularly vulnerable to complement-mediated injury, uncontrolled complement activation can lead to glomerular injury in multiple kidney diseases and end-stage renal disease.[53][54][55] Several rare types of glomerulonephritis, including dense deposit disease (DDD) and C3 glomerulonephritis (C3GN), have been classified under the umbrella term C3 glomerulopathy (C3G).[56] This group of kidney diseases is caused by abnormal control of complement activation with deposition of complement component C3 in glomeruli, resulting in glomerular damage and presenting with features of the nephritic and nephrotic syndromes.[57] C3 and C5 nephritis factors (C3Nef), which are C3-convertase stabilising antibodies, can be identified in 40% to 60% of cases of C3GN and 80% to 90% of cases of DDD.[58] Some of these patients also develop antibodies against the C5 convertase (C5Nef) and occasionally also against the CP C3 convertase (C4Nef).[59][60]
Patients with C3G, and in particular those with DDD, often show low CH50, AH50, and C3 as well as elevated levels of activation products (C3a, C3d, sC5b-9).
Atypical haemolytic uraemic syndrome (aHUS) is characterised by microangiopathic haemolytic anaemia, thrombocytopenia, and acute renal failure.[61][62] Most cases are associated with mutations or autoantibodies leading to dysregulated complement activation.[28][62]
Occasionally mutations of complement genes are also found in diarrhoea-positive HUS, caused by Shiga toxin-producing Escherichia coli.[62] Patients with atypical HUS may have a positive family history for atypical HUS.
Diagnosis includes the analysis of total haemolytic activity (CH50, AH50), C3, C3 activation products (C3a or C3d), and sC5b-9, as well as the molecular genetic analysis of C3, CFB, CFH, CHFR1-5, CFI, MCP/CD46, and THBD/CD141. Reduced C3 and normal C4 levels may already point to alternative pathway defects such as factor H and I deficiencies or autoantibodies (e.g., C3 NeF, anti-factor H) in various forms of renal diseases (e.g., aHUS, DDD).
Age-related macular degeneration
Dysregulation of the alternative pathway of the complement cascade appears to have a key role also in the pathogenesis of age-related macular degeneration (AMD).[63]
Various complement proteins, their activation products, and regulators have been identified in retinal deposits of people with AMD. Pathology-inducing polymorphisms in genes encoding complement system proteins, especially regulator factor H, as well as C3, C2, factor B, and factor I are associated with AMD.[64]
Paroxysmal nocturnal haemoglobinuria
Paroxysmal nocturnal haemoglobinuria (PNH) is a rare, acquired condition typically presenting in adults (females more than males) with the triad of haemolysis, thrombosis, and pancytopenia. It is a clonal disorder of red cells caused by mutations in the PIGA gene.[65] As a result of defective PIGA function, affected red cells lack all glycosylphosphatidylinositol-linked membrane proteins, including CD55 and CD59. The absence of CD59 and CD55 renders PNH red cells susceptible to autologous C-mediated lysis with consequent haemolytic anaemia.[66] See Paroxysmal nocturnal haemoglobinuria.
Isolated CD55 deficiency is rare but has been seen in patients with severe early-onset protein-losing enteropathy.[67] Severe Guillain-Barré syndrome-like neurological symptoms, besides haemolysis, are the hallmark symptoms of isolated CD59 deficiency.[68]
Cryoglobulinaemia
Cryoglobulinaemia can occur due to haematological malignancy. Cryoglobulins induce classical pathway activation with reduced complement C4.[69]
Partial lipodystrophy
Partial lipodystrophy is often associated with the autoantibody C3 nephritic factor. Stabilisation of C3 convertase leads to continuous complement activation and lysis of adipocytes.[70][71]
Tests for a possible complement deficiency should be performed on patients with:
Rheumatological disease (e.g., SLE and other autoimmune connective tissue diseases)
Angio-oedema without urticaria
Renal disease
Infectious disease (e.g., atypical or recurrent bacterial infection, such as cases with the rarer meningococcal serotypes and cases with recurrent meningococcal sepsis)
Partial lipodystrophy
Cryoglobulinaemia
Family history of complement deficiency.
Analysis of a potential complement deficiency should start with functional screening of each activation pathway, proceeding with identification of the defect at functional, protein, and molecular levels.[15][72][73] Given the importance of complement proteins in a wide range of biological systems, effective, standardised, and accessible tests are needed.[74]
There is great variation in quality between laboratories with regards to complement testing, and only a few tests are commercially available.[74] However, some specialised laboratories provide a comprehensive diagnosis beyond the traditional C3 and C4 parameters. European diagnostic complement labs Opens in new window
Blood samples must be separated into serum and EDTA plasma. Serum is sufficient for the analysis of the total function, complement proteins and regulators, and autoantibodies. EDTA is absolutely mandatory for the quantification of activation products. EDTA at ≥10 mM final concentration is used as standard anticoagulant because its Mg²+ and Ca²+ complexing properties block in vitro activation of the complement system.[29] Heparinised and citrated blood is less useful. If serum/plasma samples are not analysed within a few hours, they should be deep frozen -70ºC (-94ºF) and taken by courier on dry ice to a specialised laboratory. They should not undergo repeated freeze-thaw cycles due to the risk of in vitro activation.
Global tests, such as the haemolytic assays for the classical pathway (CH50) and the alternative pathway (AH50), or a liposome-based CP lysis assay, provide information about how the whole complement cascade is working. These assays artificially measure target cell lysis in test tubes. The tests are dependent on the full assembly of membrane attack complexes (C5b-C9) to cause target cell lysis. A missing, or considerably reduced, activity indicates a primary complement deficiency. However, this finding may also be due to increased complement consumption leading to secondary deficiency.[29] Reduced C3 and normal C4 levels are indicative of alternative pathway (AP) regulator defects, such as factor H and I deficiencies, or C3 nephritic factor-induced AP dysregulation, as seen in various forms of nephropathies (e.g., atypical haemolytic uraemic syndrome [aHUS], C3 glomerulopathy [C3G]/dense deposit disease [DDD]). Normal C3 and C4 levels do not exclude cryoglobulinaemia. Serum cryoglobulin levels should be tested when clinical suspicion of diseases associated with reduced complement C4 is high (e.g., when patients present with vasculitic rash, particularly affecting the colder extremities; arthritis; and/or acute renal failure). The definitive diagnostic test involves obtaining fresh clotted blood, collected in a prewarmed thermos flask (at 37ºC [98.6ºF]); serum is then rapidly separated to reduce the risk of cryoglobulin being concealed within clotted blood.
For rapid deficiency analysis, an enzyme-linked immunosorbent assay (ELISA) has been developed that examines all three activation pathways in parallel.[75] Individual analysis of components is subsequently required to determine in which portion of the complement cascade defects occur. Analysis of complement activation products such as C3a, C3d, or sC5b-9 is necessary to distinguish primary from secondary complement deficiency due to over-activation and is helpful for determining the efficacy of complement-targeted therapy (e.g., by eculizumab).[76]
Molecular testing has become essential to elucidate the genetic background of complement deficiencies and is routinely included in diagnostics of, for example, nephropathies and hereditary angio-oedema.[28][77]
Theoretically, deficient complement proteins can be replaced, for instance using fresh frozen plasma (FFP) from blood donors. In practice this is not generally recommended, because the half-life of infused complement proteins is too short and the proteins may be pro-inflammatory. Despite these issues, FFP has benefitted some patients (e.g., patients with atypical haemolytic uraemic syndrome [aHUS]).[78] In cases of severe dysregulation, as seen in paroxysmal nocturnal haemoglobinuria and severe renal disorders such as aHUS and C3 glomerulopathy, complement inhibitors such as eculizumab and ravulizumab (complement C5 inhibitors), or pegcetacoplan (a complement C3 inhibitor) have been successful.[79][80][81][82] Certain vaccinations may be required before starting these treatments.
Therapy of the various forms of angio-oedema has meanwhile gone far beyond C1-INH replacement, including drugs that directly target the bradykinin receptor (e.g., icatibant) and drugs that target plasma kallikrein (e.g., ecallantide, sebetralstat).[83][84] Sebetralstat demonstrated rapid suppression of kallikrein activity and faster symptom relief as compared to placebo in a phase 2 trial, leading to initiation of phase 3 evaluation.[85]
Antibiotic prophylaxis may be considered for patients with recurrent infections despite appropriate vaccination, particularly those with high exposure risk (e.g., living in endemic areas or working in high-risk professions). For others, ensuring access to emergency antibiotics and prompt medical review should form part of the emergency plan. The decision between prophylactic and emergency antibiotics should be individualised, balancing potential benefits with the risk of antibiotic resistance.[15]
Generic clinical management steps include:[15]
Screening relatives for occult complement deficiency
Encouraging and optimising immunisation programmes (seeking to boost protective antibody levels where possible). The same vaccines are recommended for patients with complement deficiencies as for healthy individuals. Some vaccines of particular focus (e.g., pneumococcal, meningococcal, and Hib vaccines) are indicated for use outside the routinely recommended age because of the increased risk of vaccine-preventable disease in this population.[15][51][52][86]
Considering prescription of preventive daily (lifelong) antibiotics (similar to antibiotic prophylaxis post-splenectomy).
Annual follow-up to provide patient education, guidance on vaccination, antibiotics when required, emergency management advice, and family studies if indicated.
A number of organisations and professional societies have produced guidelines to assist healthcare providers in the diagnosis and management of complement deficiencies and their associated features. These include:
European Society for Immunodeficiencies (ESID) and European Reference Network on Rare Primary Immunodeficiency, Autoinflammatory and Autoimmune Diseases (ERN RITA). ESID and ERN RITA complement guideline: deficiencies, diagnosis, and management.[15]
Guideline describing the complement system and the functions of the constituent pathways, with particular focus on primary immunodeficiencies and their diagnosis and management.
World Allergy Organization (WAO) and the European Academy of Allergy and Clinical Immunology (EAACI). The international WAO/EAACI guideline for the management of hereditary angioedema - the 2021 revision and update.[35]
Global guideline on the diagnosis and management of HAE, including key recommendations and guidance on common and important clinical issues in patients with HAE with deficient C1 inhibitor (type 1) and HAE with dysfunctional C1 inhibitor (type 2). The guideline aims to help establish global standards for HAE management and to encourage and facilitate the use of recommended diagnostics and therapies for all patients.
The Advisory Committee on Immunization Practices (ACIP), Centers for Disease Control and Prevention (CDC). CDC: adult immunization schedule by age Opens in new window
The adult immunisation schedule summarises ACIP adult immunisation recommendations, as well as listing the contraindications to and precautions for all routinely recommended vaccines in the schedule. It also describes the recommended adult immunisation schedule by medical condition, including complement deficiencies.
The Advisory Committee on Immunization Practices (ACIP), Centers for Disease Control and Prevention (CDC). CDC: child and adolescent immunization schedule by age Opens in new window
The child and adolescent immunisation schedule summarises ACIP child and adolescent immunisation recommendations, as well as listing the contraindications to and precautions for all vaccine types in the schedule. It also describes the recommended child and adolescent immunisation schedule by medical condition, including complement deficiencies.
The Advisory Committee on Immunization Practices (ACIP), Centers for Disease Control and Prevention (CDC). General best practice guidelines for immunization: altered immunocompetence.[86]
Guideline discussing general best practice for immunisation in patients with altered immunocompetence, including indications for administering a vaccine outside of routinely recommended age groups.
C-1-INH Deficiency Working Group. International consensus and practical guidelines on the gynecologic and obstetric management of female patients with hereditary angioedema caused by C1 inhibitor deficiency.[87]
Guidelines for optimising the management of gynecological and obstetric events in female patients with HAE caused by C1-INH deficiency.
International Complement Society (ICS) and the International Union of Immunological Societies (IUIS). Complement analysis 2016: clinical indications, laboratory diagnostics and quality control.[72][74]
Guidelines on modern complement analysis and standards for the development of international testing programmes.
Use of this content is subject to our disclaimer