Aetiology
Pemphigus vulgaris (PV) and pemphigus foliaceus (PF) are autoimmune, blistering skin diseases that are antibody mediated. PV accounts for around 70% of pemphigus cases, and PF accounts for around 20%.[17]
Paraneoplastic pemphigus (PNP) is both autoimmune (antibody-mediated) and cell-mediated; it accounts for around 5% of cases.[10][12] Pemphigus erythematosus and IgA pemphigus are rare variants.
Pemphigus is a group of IgG-mediated autoimmune diseases of stratified squamous epithelia, such as the skin and oral mucosa, in which acantholysis (the loss of cell adhesion) causes blisters and erosions.
In PV and PF, IgG autoantibodies bind exclusively to the desmoglein component (a cadherin-like adhesion molecule) of the desmosome (a protein complex responsible for adhesion between skin cells). The autoantibodies, characteristically against desmoglein 3 (and to a lesser extent, desmoglein 1) in PV and desmoglein 1 in PF, cause disruption of cell attachment by binding the extracellular portion of the keratinocyte.[5] In PNP, the autoantibodies bind portions of the hemidesmosome, resulting in more complex pathophysiology.[18][19]
Most cases of pemphigus develop spontaneously. Rarely, PV and PF may be associated with exposure to certain drugs (e.g., penicillamine, tiopronin, ACE inhibitors, penicillin, interleukin-2, nifedipine, and rifampicin). The geographical distribution of endemic PF parallels that of the black fly, which is thought to be a possible vector that precipitates the disease.[20][21] Other haematophagous insects, such as bedbugs and kissing bugs, have also been implicated.[22] In endemic PF, desmoglein 1 antibodies have been found to cross-react with sand fly salivary LJM11 antigen.[23][24]
PNP is strongly linked to underlying malignancies. In one third of patients, PNP is a marker for unknown malignancy. The distribution of PNP is bimodal, meaning a disease that occurs in young people, typically associated with non-human herpesvirus 8 Castleman's disease, and in older people with a history of non-Hodgkin's lymphoma, chronic lymphocytic leukaemia, thymoma, and rare sarcomas.[4]
Pathophysiology
Pemphigus is a group of IgG-mediated autoimmune diseases of stratified squamous epithelia; primarily presenting in skin and oral mucosa. Pemphigus vulgaris (PV) is the most common subtype.
Both pemphigus vulgaris and pemphigus foliaceous are characterised by autoantibodies to desmoglein (anti-desmoglein 3, and to a lesser extent, anti-desmoglein 1, in PV; anti-desmoglein 1 in PF). Desmoglein 1 and 3 are both expressed in the mouth and skin.
Distribution and expression levels of desmoglein 1 and desmoglein 3 may account for the characteristic distribution and localisation of disease in patients with PV or PF. Patients with PV (i.e., with anti-desmoglein 3 autoantibodies) develop mucosal disease because there is insufficient expression of desmoglein 1 to maintain mucosal keratinocyte adhesion and prevent oral erosions. A subset of patients with PV do not develop skin disease, as the prominent expression of desmoglein 1 in the skin is sufficient to stabilise keratinocyte adhesion and prevent blistering. However, many patients with PV develop autoantibodies to desmoglein 1, leading to more severe skin disease.[25] The underlying antibody profile is, therefore, an important determinant of PV phenotype.
In patients with PF who have pathological anti-desmoglein 1 autoantibodies, the mouth is spared because the expression of desmoglein 3 can maintain mucosal keratinocyte adhesion.
Although PV and PF are categorised as distinct conditions, there is evidence to suggest that they represent a spectrum of disease. For example, pemphigus confined to the mouth may become generalised to the skin; less commonly, cases of patients with classic PF progressing to PV have been reported. In theory, this may be due to epitope spreading (an epitope is an antigenic site on a protein that stimulates an antibody response). In epitope spreading, the autoimmune process expands to recognise either chemically similar epitopes, or epitopes that are similar to the initial autoantigen.[26]
PNP has a more complex pathophysiology, but the presence of autoantibodies that prevent keratinocyte adhesion is similar to PV and PF. PNP has components both of a humoral autoimmune process (i.e., mediation by antibodies) and of a cell-mediated process (i.e., cytotoxic T cells predominate). This is reflected by disease findings that are similar to those found in cytotoxic T cell-mediated processes, such as erythema multiforme, graft-versus-host disease, and lichen planus, while also having classic features of pemphigus.[27]
Classification
There is no formal classification, but pemphigus is typically classified into three major forms.
Pemphigus vulgaris[1][3]
Pemphigus vulgaris is the most common variant.
Usually begins with oral mucosal lesions (buccal and/or gingival); painful, persisting erosions that interfere with eating. Less commonly, ocular, nasal, laryngeal, oesophageal, genital, and anal erosions are seen.
Skin involvement presents with flaccid blisters with clear content. Blisters develop on non-erythematous skin, quickly transforming into postbullous erosions.
Blisters and erosions may be localised or generalised, and predominate at seborrhoeic areas (chest, face, scalp, interscapular region) and on the extremities.
Routine histology demonstrates acantholysis (loss of cell-cell adhesion) superior to the basement membrane of the skin in the lower portion of the epidermis, often with hair follicle involvement.
Presence of autoantibodies against desmoglein 3 (Dsg3), and to a lesser extent, desmoglein 1 (Dsg1).[3]
[Figure caption and citation for the preceding image starts]: Erosions of the oral mucosa in a patient with pemphigus vulgarisImage courtesy of Dr Jon Meyerle [Citation ends].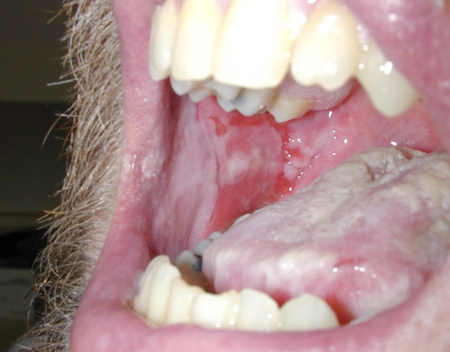 [Figure caption and citation for the preceding image starts]: Widespread erosions and scattered intact blisters in a patient with widespread pemphigus vulgarisImage courtesy of Dr Jon Meyerle [Citation ends].
[Figure caption and citation for the preceding image starts]: Widespread erosions and scattered intact blisters in a patient with widespread pemphigus vulgarisImage courtesy of Dr Jon Meyerle [Citation ends].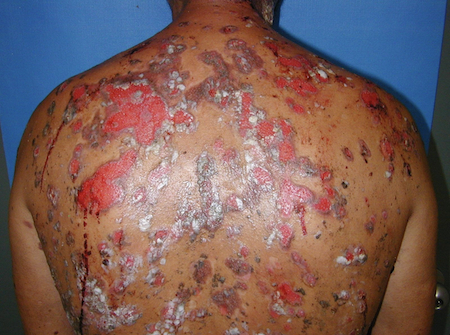
Pemphigus foliaceus[1]
Skin involvement: transient, flaccid blisters or crusty erosions in seborrhoeic skin areas (chest, scalp, face, interscapular region).
More extensive skin involvement in sporadic and endemic pemphigus foliaceus ('fogo selvagem', Brazilian pemphigus, Tunisian pemphigus).
No mucosal involvement.
Routine histology sometimes demonstrates acantholysis at the level of the granular cell layer, but often the stratum corneum (top portion of the epidermis) is lost.
Presence of autoantibodies against desmoglein 1 (Dsg1).
[Figure caption and citation for the preceding image starts]: Pemphigus foliaceusImage courtesy of Dr Jon Meyerle [Citation ends].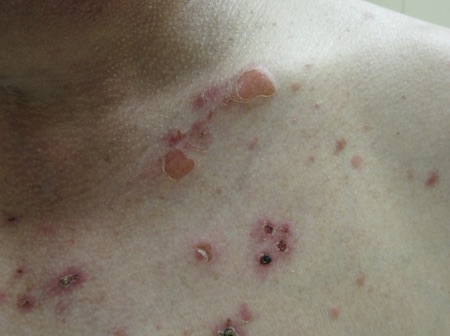 [Figure caption and citation for the preceding image starts]: Pemphigus foliaceusImage courtesy of Dr Jon Meyerle [Citation ends].
[Figure caption and citation for the preceding image starts]: Pemphigus foliaceusImage courtesy of Dr Jon Meyerle [Citation ends].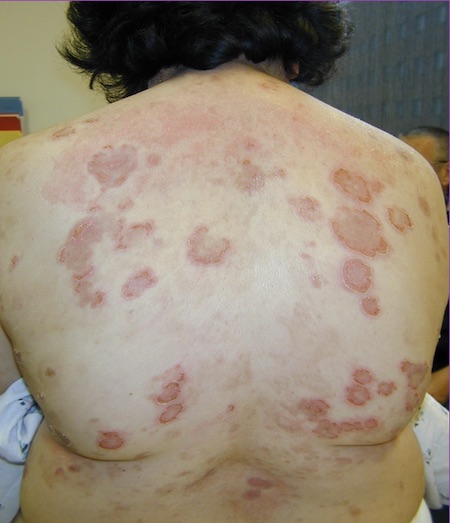
Paraneoplastic pemphigus[4][5]
Presents in the context of concomitant malignancy, particularly non-Hodgkin's lymphoma, chronic lymphocytic leukaemia, thymoma, or Castleman’s disease. The underlying malignancy may not have been diagnosed at the time of pemphigus diagnosis.
Mucosal involvement: initially limited cheilitis and/or ulcerative stomatitis, persisting painful erosions that lead to severe dysphagia. Cicatricial conjunctivitis, keratitis, and genital involvement are common. Pharyngeal involvement is possible; the nasal cavity and oesophagus can also be affected.
Cutaneous polymorphic lesions with symptoms resembling mild lichen planus-like to graft-versus-host disease-like, erythema multiforme-like, bullous pemphigoid-like, or pemphigus vulgaris-like eruption. Palmar involvement is common.
Pulmonary involvement (alveolitis, bronchiolitis obliterans, pulmonary fibrosis) is a characteristic and life-threatening complication.
Histopathology findings include:
Epidermal acantholysis, dyskeratosis, and vacuolar interface changes
Epidermal intercellular deposition of IgG and C3, with or without linear deposition at the basement membrane zone as demonstrated by direct immunofluorescence staining
Serum autoantibodies to epithelia as demonstrated by indirect immunofluorescence with murine tissue (bladder, heart, liver, and tongue)
Autoantibodies to several cytoplasmic proteins of the plakin family (epiplakin, plectin, desmoplakin I, desmoplakin II, BPAG1 [also known as dystonin], envoplakin, and periplakin).
[Figure caption and citation for the preceding image starts]: Patient with paraneoplastic pemphigusImage courtesy of Dr Jon Meyerle [Citation ends].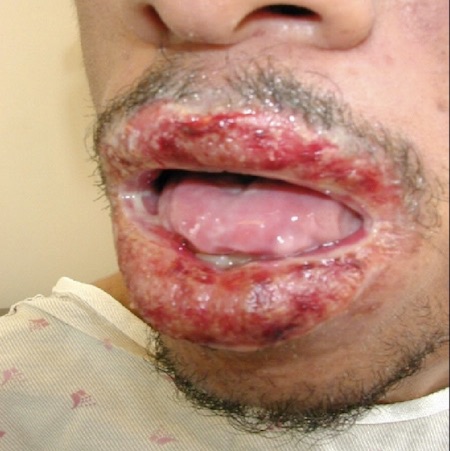
Use of this content is subject to our disclaimer